MAE. Existuje mnoho odlišných dětství epilepsie syndromy že jsme se stali kriticky vědom v genomické éře, jak jsou spojeny s významnými genetické příčiny, včetně Dravet Syndrom (SCN1A) a Epilepsie Dětství s Migrující Fokální Záchvaty (KCNT1). Existuje však mnoho dalších epilepsických syndromů, kde je genetická příčina již dlouho podezřelá, ale zůstala nepolapitelná. Jeden z epilepsie syndromy, které se do značné míry zůstává neprozkoumané je Dooseho Syndrom, označovaný také jako Astatic Myoklonické Epilepsie (MAE) nebo Epilepsie s Myoklonickými-Atonické Záchvaty. V nedávné studii v Epilepsia, jsme zkoumali genetické architektury Dooseho Syndrom a identifikovány monogenních příčin u 14% jedinců, včetně SYNGAP1, NEXMIF (KIAA2022), a SLC6A1. Naše studie naznačuje, že Dooseův syndrom je geneticky heterogenní, možná s odlišnou genetickou krajinou.
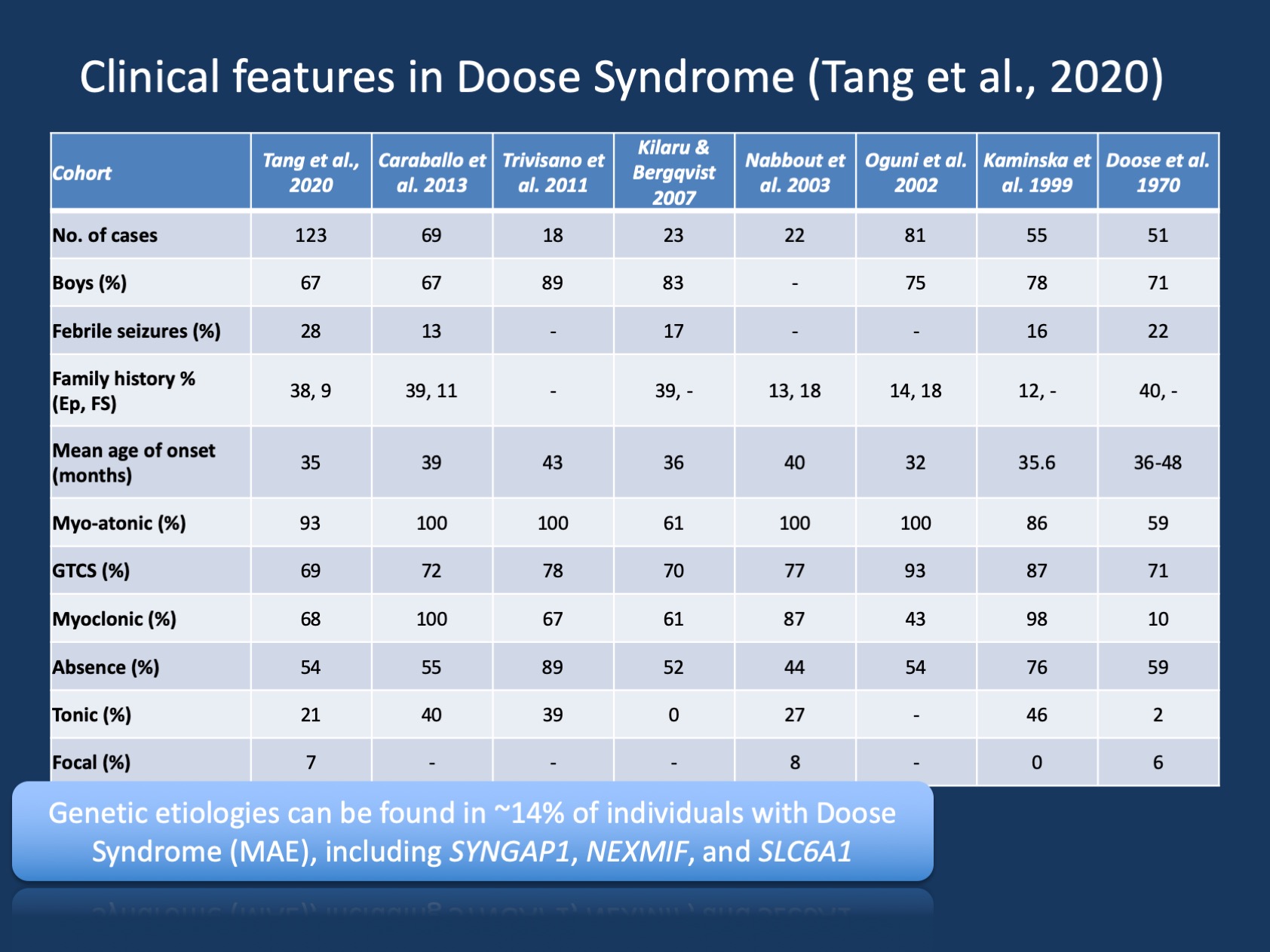
Obrázek 1. Klinické příznaky Dooseho syndromu (epilepsie s myoklonickými atonickými záchvaty nebo myoklonickou Astatickou epilepsií). Tabulka je převzata z naší nedávné publikace Tang et al., 2020 a porovnává klinické rysy ve velkých kohortách Dooseho syndromu, které byly hlášeny od počátečního popisu v roce 1970. Dooseho syndrom (MAE) je vzácný epilepsický syndrom, ale je rozpoznatelnou klinickou entitou. Na rozdíl od Dravetova syndromu, kde >90% jedinců má varianty způsobující onemocnění v SCN1A, je Dooseův syndrom geneticky heterogenní.
historie MAE. Nejprve mi dovolte vyjasnit některé problémy s pojmenováním. Dooseho Syndrom nebo Astatic Myoklonické Epilepsie (MAE) je nyní oficiálně označovány jako Epilepsie s Myoklonickými-Atonické Záchvaty pomocí nejnovější pojmenování. Nicméně, zatímco se obvykle snažíme sladit s oficiálním způsobem, jak se používají termíny související s epilepsií, dovolte mi, abych zde byl trochu tvrdohlavý. Jako dítě neurologa vyškoleni v Oddělení Neuropediatrics v Kiel, Německo, budu odkazovat na tento stav jako Dooseho Syndrom pomocí MAE jako zkratka, ctít Hermann Doose (1927-2018), který je jedním ze zakladatelů Evropské pediatrické epileptologii (odkaz na ILAE in memoriam). Mnoho rodilých mluvčích angličtiny obvykle špatně vyslovuje výslovnost-správná výslovnost je „Dohs-ah“ a nerýmuje se s „husou“.
klinický obraz MAE. Doose je obvykle připočítán s důležitým klinickým pozorováním: v roce 1960, on a jeho spolupracovníci v Kielu uznávané neobvyklé epilepsie u dětí, které se vyznačuje náhlým nástupem drop attacks (atonické záchvaty), myoklonické záchvaty, generalizované tonicko-klonické záchvaty, generalizované EEG vzor, obvykle se vyskytující ve věku mezi dvěma a pěti. Tato epilepsie se obvykle vyskytovala u dětí s předchozím typickým vývojem (viz níže) a často vedla k vývojovému zpoždění a mentálnímu postižení. Doose a spolupracovníků zřejmé, že tato nově uznané epilepsie syndromu se liší od Lennox-Gastautovým Syndromem nebo epilepsií generalizované tonicko-klonické záchvaty způsobené horečkou, latter který byl následně pojímány jako Dravet Syndrom.
porozumění MAE. Klinické popisy Dooseho Syndrom (MAE) byla stabilní v průběhu let, ale MAE se nedostalo tolik pozornosti jako dětství epilepsie syndromy, kde je genetický základ byl objeven a kde cílený výzkum může umožnit nové způsoby léčby. V naší nedávné publikaci Tang and collaborators jsme uvedli osm studií za posledních 50 let mezi lety 1970 a 2020 s celkem 442 popsanými jednotlivci. Proto je MAE vzácná, ale je dost běžné, že většina dětských neurologů a dětských epileptologů si je tohoto stavu vědoma. Pouze malá podskupina jedinců s MAE má strukturální nálezy mozku nebo metabolické abnormality, které vysvětlují základní epilepsii – u velké většiny dětí s MAE, neuroimaging a metabolická práce jsou všední.
nalezení příčiny MAE. Absence vysvětlujících zobrazovacích nebo metabolických nálezů vyvolává obavy ze základní genetické příčiny. Zatímco předchozí studie se pokusil pin na genetické příspěvek prostřednictvím epidemiologických studií o posuzování rodinné historie, tato tradiční metoda byla do značné míry nahrazena nedávné objevy v epilepsie genetiky. Posouzení genetického základu těžkých dětských epilepsií se nyní provádí spíše molekulární genetikou než genetickou epidemiologií. Posouzení genetické architektury MAE bylo jedním z cílů naší studie Tang a spolupracovníků, která byla publikována v epilepsii.
je MAE jednou nemocí? Před popisem genetických nálezů v MAE, dovolte mi představit jednu probíhající debatu v oblasti MAE. Počáteční popis Doose a spolupracovníků do značné míry zahrnoval děti s typickým vývojem před nástupem záchvatů (>75%), což je srovnatelné s naší nedávnou studií (79%). Nicméně, při srovnání dětí s a bez předchozí vývojové problémy, jsme při pohledu na stejnou nemoc, nebo by měly pouze děti se před všední rozvoj, být považovány za typické MAE? I když tato otázka není snadno vyřešena při zvažování historie a výsledků epilepsie, základní genetické změny mohou poskytnout určité stopy. Genetické příčiny jsou identifikovány častěji u dětí s těžšími poruchami neurologického vývoje a všechny děti s identifikovaným genetické příčiny měl komplex vývojových poruch, kromě epilepsie. Proto, genetické příčiny mohou být pozorovány u dětí s MAE, ale obvykle pouze u dětí s MAE a komorbidní poruchy neurologického vývoje, což naznačuje, že MAE je nejen geneticky heterogenní stav, ale má podobné vlastnosti jako jiné epilepsie v dětství s ohledem na jejich genetickou heterogenitu a komorbidity. Například, v Západní Syndrom (Infantilní Spazmy), genetické příčiny jsou také zjišťovány pouze u dětí s vývojovými zjištěních, a ne u dětí, které přímo reagují na léky bez neurologického následky.
genetické nálezy u MAE. Genetické spektrum u MAE je jiné než u epileptických encefalopatií obecně. Genetické testování bylo provedeno u 85 jedinců a 12/85 (14%) jedinců mělo pozitivní genetické nálezy. Tři geny byly nalezeny u dvou jedinců, včetně SYNGAP1, NEXMIF (KIAA2022) a SLC6A1. Tato trojice genů byla dříve spojena s vývojovými a epileptickými encefalopatiemi s generalizovanými záchvaty. Kromě toho byli jednotlivci nalezeni s variantami způsobujícími onemocnění v KCNA2, SCN2A, STX1B, KCNB1 a MECP2. Všechny tyto geny byly dříve diskutovány na našem blogu a je známo, že mají generalizované záchvaty a generalizované funkce EEG.
chybí geny v MAE. Zatímco některé předchozí zprávy našly varianty v SCN1A, toto nebylo nalezeno v naší kohortě. Kromě toho, jiné běžné epilepsie geny jsou pozoruhodně chybí, včetně CDKL5 a SLC2A1 (GLUT1). To naznačuje, že genetická Krajina MAE je poněkud odlišná a odlišná od jiných vývojových a epileptických encefalopatií, i když z relativně malých kohort lze vyvodit pouze omezené závěry. Diagnostický výtěžek u MAE není příliš vysoký-14% je mnohem nižší, než je obvykle pozorováno u vývojových a epileptických encefalopatií. Při vyloučení jedinců s typickým vývojem jsou však diagnostické výnosy mnohem vyšší, což naznačuje, že genetické testování má hodnotu u dětí s MAE. Obecně, i když žádný jednotlivec s MAE neměl varianty způsobující onemocnění u SLC2A1, genetické testování léčitelných příčin genetických epilepsií může být obecně zvažováno u atypických generalizovaných epilepsií.
chybějící dědičnost MAE. Pokud je genetické testování negativní pro MAE u více než 80% jedinců, kde je skrytá genetická zátěž? Od roku 2020 bych navrhoval dvě možná vysvětlení. Za prvé, genetická zátěž generalizovaných epilepsií není zcela neznámá. Je vysvětleno více než 30% populačního rizika pro stavy, jako je epilepsie v dětství (CAE) nebo juvenilní myoklonická epilepsie (JME). Toto vysvětlení však není dáno jednogenovými příčinami, ale polygenním rizikem s odkazem na aditivní účinek tisíců běžných genetických variant. Lze předpokládat, že MAE může představovat „extrémní fenotyp“ generalizovaných epilepsií, kde je toto polygenní riziko zvláště vysoké. Tato hypotéza je testovatelná a bude určitě sledována v budoucnu. Kromě toho je v lidském genomu více než exom-nedávné nálezy v epilepsiích zdůraznily roli nekódujících variant, jako jsou opakované expanze v familiární dospělé myoklonické epilepsii (FAME). Vzhledem k nápadně podobné klinické rysy v mnoha dětí s MAE, to může ještě být rozumné hledat společnou genetickou příčinu pro mnoho dětí s MAE mimo exome, pomocí sekvenování celého genomu. Užívání spolu, posuzování rizika polygenní skóre (VAS) a sekvenování celého genomu (WGS) jsou dvě potenciální cesty k další identifikaci genetického základu MAE.
co potřebujete vědět. Dooseho syndrom (MAE) je pravděpodobně genetický epilepsický syndrom s heterogenní příčinou. Nemoc-působit varianty jsou přítomny v 14% dětí a jsou obvykle zjištěny u dětí s dalšími vývojovými rysy, jako jsou vývojové zpoždění nebo autismus. Trojice genů včetně SYNGAP1, NEXMIF (KIAA2022), a SLC6A1, jsou opakující se genetické příčiny, které mohou narážet na sdílené základní biologie, který je poněkud odlišný od jiných vývojových a epileptických encefalopatií.

Ingo Helbig je dětský neurolog a výzkumník genetiky epilepsie pracující v dětské nemocnici ve Filadelfii (CHOP) v USA. Vede také skupinu epilepsie genetics group na univerzitě v Kielu v Německu.

