MAE. On olemassa monia erillisiä lapsuuden epilepsiaoireyhtymiä, jotka olemme kriittisesti tietoisia genomien aikakaudella, koska ne liittyvät näkyvästi geneettisiin syihin, mukaan lukien Dravetin oireyhtymä (SCN1A) ja lapsenkengissä oleva epilepsia, jolla on siirtyviä Polttovälikohtauksia (KCNT1). On kuitenkin monia muita epilepsiaoireyhtymiä, joissa geneettistä syytä on pitkään epäilty, mutta se on jäänyt vaikeasti havaittavaksi. Yksi epilepsiaoireyhtymistä, joka on suurelta osin jäänyt tutkimatta, on Doose-oireyhtymä, jota kutsutaan myös Myokloniseksi Astaattiseksi Epilepsiaksi (MAE) tai Epilepsiaksi Myoklonis-Atonisilla kohtauksilla. Tuoreessa Epilepsiatutkimuksessa selvitimme Doosen oireyhtymän geneettistä arkkitehtuuria ja tunnistimme monogeeniset syyt 14%: lla yksilöistä, mukaan lukien SYNGAP1, NEXMIF (KIAA2022) ja SLC6A1. Tutkimuksemme viittaa siihen, että Doosen oireyhtymä on geneettisesti heterogeeninen, mahdollisesti omanlaisensa geneettinen maisema.
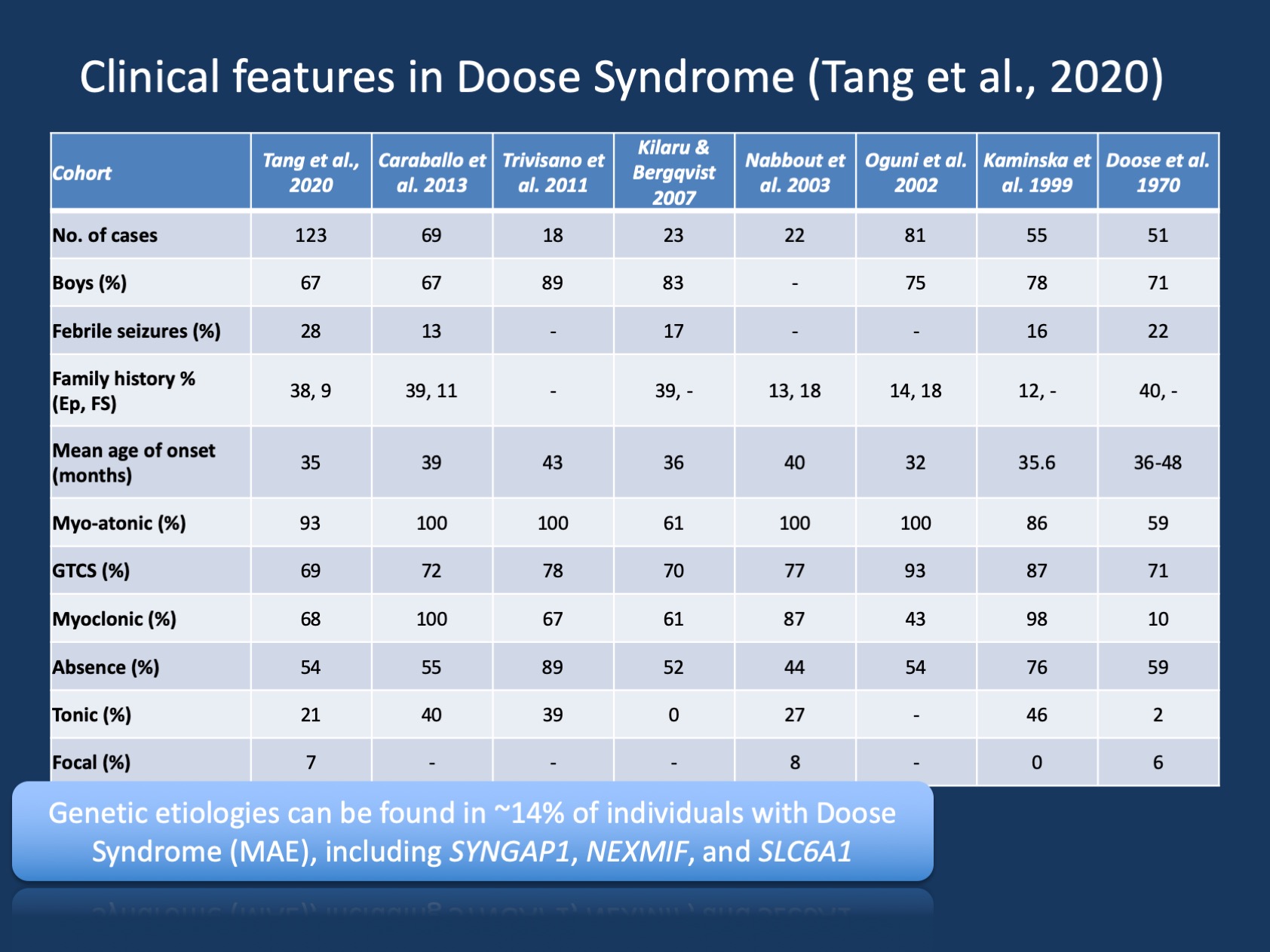
Kuva 1. Doose-oireyhtymän kliiniset piirteet (myoklonisiin atonisiin kohtauksiin liittyvä epilepsia tai myokloninen Astaattinen epilepsia ). Taulukko on mukautettu meidän tuore julkaisu Tang et al., 2020 ja vertaa doosen oireyhtymän suurten kohorttien kliinisiä ominaisuuksia, joita on raportoitu ensimmäisen kuvauksen jälkeen vuonna 1970. Doosen oireyhtymä (MAE) on harvinainen epilepsiaoireyhtymä, mutta se on tunnistettava kliininen kokonaisuus. Toisin kuin Dravetin oireyhtymässä, jossa >90 prosentilla yksilöistä on scn1a: n sairauksia aiheuttavia variantteja, Doosen oireyhtymä on geneettisesti heterogeeninen.
Maen historia. Selvitän ensin muutamia nimeämisasioita. Doose-oireyhtymää tai myoklonista Astaattista epilepsiaa (Mae) kutsutaan nyt virallisesti Myoklonis-Atonisiksi kohtauksiksi käyttäen viimeisintä nimeämiskäytäntöä. Vaikka yritämme yleensä noudattaa virallista tapaa, jolla epilepsiaan liittyviä termejä käytetään, sallikaa minun olla hieman itsepäinen tässä asiassa. Kielissä Saksassa Neuropediatrian laitoksella koulutettuna lastenneurologina viittaan tähän sairauteen DOOSEN oireyhtymänä käyttäen MAE: ta pikakirjoituksena, kunnioittaen Hermann Doosea (1927-2018), joka on yksi Euroopan lastentautien epileptologian perustajista (linkki ilae in memoriamiin). Monet äidinkielenään englantia puhuvat saavat tyypillisesti ääntämyksen väärin-oikea ääntämys on ”Dohs-ah” eikä rimmaa ”Goosen”kanssa.
mae: n kliininen kuva. Doose on tyypillisesti hyvitetään tehdä tärkeä kliininen havainto: 1960-luvulla hän ja hänen yhteistyökumppaninsa kielissä tunnistivat lapsilla epätavallisen epilepsian, jolle oli ominaista äkilliset pudotuskohtaukset (atoniset kohtaukset), myokloniset kohtaukset, yleistyneet toonis-klooniset kohtaukset ja yleistynyt EEG-kuvio, joka esiintyy tyypillisesti kahden ja viiden vuoden iässä. Tätä epilepsiaa esiintyi tyypillisesti lapsilla, joilla oli aiemmin tyypillistä kehitystä (KS.jäljempänä) ja se johti usein kehityksen viivästymiseen ja kehitysvammaisuuteen. Doose ja yhteistyökumppanit ymmärsivät, että tämä vastikään tunnustettu epilepsiasyndrooma oli erilainen kuin Lennox-Gastaut-oireyhtymä tai epileptiset kohtaukset, joihin liittyi yleistyneitä toonis-kloonisia kohtauksia, jotka saivat alkunsa kuumeesta, ja joista jälkimmäinen myöhemmin käsitettiin Dravetin oireyhtymäksi.
ymmärrys MAE. Doosen oireyhtymän (MAE) kliiniset kuvaukset ovat olleet tasaisia vuosien varrella, mutta Mae ei ole saanut yhtä paljon huomiota kuin lapsuuden epilepsiaoireyhtymät, joissa on löydetty geneettinen perusta ja joissa Kohdennettu tutkimus voi mahdollistaa uusia hoitoja. Tang and collaboratorsin tuoreessa julkaisussa listasimme kahdeksan tutkimusta viimeisten 50 vuoden ajalta vuosina 1970-2020, joissa oli yhteensä 442 kuvattua henkilöä. Näin ollen MAE on harvinainen, mutta se on tarpeeksi yleistä, että useimmat lasten neurologit ja lasten epileptologit ovat tietoisia tästä tilasta. Vain pienellä osalla MAE-potilaista on rakenteellisia aivolöydöksiä tai metabolisia poikkeavuuksia, jotka selittävät taustalla olevan epilepsian – valtaosalla MAE-tautia sairastavista lapsista neuroimagointi ja metabolinen tutkimus ei ole merkittävä.
MAE: n syyn löytäminen. Selittävien kuvantamis-tai aineenvaihduntalöydösten puuttuminen herättää huolta taustalla olevasta geneettisestä syystä. Vaikka aiemmissa tutkimuksissa oli pyritty osoittamaan geneettistä osuutta epidemiologisten tutkimusten avulla arvioimalla sukuhistoriaa, tämä perinteinen menetelmä on suurelta osin syrjäyttänyt viimeaikaiset löydöt epilepsiagenetiikassa. Vaikeiden lasten epileptikkojen geneettisen perustan arviointi tehdään nykyään molekyyligenetiikan eikä geneettisen epidemiologian avulla. Maen geneettisen arkkitehtuurin arviointi oli yksi tavoitteista Tangin ja yhteistyökumppaneiden tekemässä tutkimuksessa, joka julkaistiin Epilepsia-lehdessä.
onko MAE yksi sairaus? Ennen kuin kuvailen mae: n geneettisiä löydöksiä, haluan esitellä yhden käynnissä olevan keskustelun MAE: n alalla. Doose ja yhteistyökumppanit kuvasivat aluksi pääasiassa lapsia, joilla oli tyypillinen kehitys ennen kohtausten puhkeamista (>75%), mikä on verrattavissa tuoreeseen tutkimukseemme (79%). Vertailtaessa lapsia, joilla on aiempia kehityshäiriöitä ja joilla ei ole niitä, tarkastelemmeko kuitenkin samaa tautia vai pitäisikö vain lapsia, joiden edeltävä kehitys ei ole huomattava, pitää tyypillisinä MAE: einä? Vaikka tämä kysymys ei ole helppo ratkaista, kun tarkastellaan epilepsia historia ja tulokset, taustalla geneettiset muutokset voivat antaa joitakin vihjeitä. Geneettiset syyt tunnistetaan useammin lapsilla, joilla on vaikeampia neurokehityshäiriöitä, ja kaikilla lapsilla, joilla on todettu geneettisiä syitä, oli epilepsian lisäksi monimutkaisia neurokehityshäiriöitä. Näin ollen geneettisiä syitä voidaan havaita lapsilla, joilla on MAE, mutta tyypillisesti vain lapsilla, joilla on MAE ja liitännäissairauksia neurokehityshäiriöt, mikä viittaa siihen, että MAE ei ole vain geneettisesti heterogeeninen tila, vaan sillä on samanlaisia piirteitä kuin muilla epilepsioilla lapsuudessa niiden geneettisen heterogeenisyyden ja samanaikaisuuden suhteen. Esimerkiksi Länsi-syndroomassa (infantiili Spasms) geneettisiä syitä havaitaan myös vain lapsilla, joilla on neurokehityslöydöksiä, eikä lapsilla, jotka reagoivat suoraan lääkitykseen ilman neurokehitysoireita.
mae: n geneettiset löydökset. Mae: n geneettinen kirjo on erilainen kuin epileptisissä enkefalopatioissa yleensä. Geenitestejä tehtiin 85 yksilölle ja 12/85 (14%) yksilöillä oli positiivisia geneettisiä löydöksiä. Kolmesta geenistä löytyi kaksi yksilöä, muun muassa SYNGAP1, NEXMIF (KIAA2022) ja SLC6A1. Tämä geenien kolmijako on aiemmin yhdistetty kehityshäiriöihin ja epileptisiin enkefalopatioihin, joihin liittyy yleistyneitä kohtauksia. Lisäksi yksittäisiltä yksilöiltä löydettiin tautia aiheuttavia variantteja luokista KCNA2, SCN2A, STX1B, KCNB1 ja MECP2. Kaikki nämä geenit aiemmin keskusteltiin blogissamme ja niiden tiedetään esittävän yleistyneitä kohtauksia ja yleistyneitä EEG-ominaisuuksia.
puuttuvat geenit MAE: ssa. Vaikka joissakin aiemmissa raporteissa on löydetty variantteja SCN1A: sta, tätä ei löytynyt kohortistamme. Lisäksi muita yleisiä epilepsiageenejä puuttuu huomattavasti, kuten CDKL5 ja SLC2A1 (GLUT1). Tämä viittaa siihen, että MAE: n geneettinen maisema eroaa jonkin verran muista kehityshäiriöistä ja epileptisistä enkefalopatioista, vaikka suhteellisen pienistä kohorteista voidaan tehdä vain rajallisia johtopäätöksiä. Mae: n diagnostinen tuotto ei ole kovin suuri – 14% on paljon pienempi kuin tyypillisesti kehityshäiriöissä ja epileptisissä enkefalopatioissa. Kuitenkin, kun jätetään pois yksilöt, joilla on tyypillinen kehitys, diagnostinen tuotto on paljon suurempi, mikä viittaa siihen, että geneettinen testaus on arvokasta lapsilla MAE. Yleisesti ottaen, vaikka yhdelläkään MAE: tä sairastavalla henkilöllä ei ollut slc2a1: ssä tautia aiheuttavia variantteja, geneettistä testausta hoidettavissa olevien geneettisten epileptikkojen syiden selvittämiseksi voidaan yleensä harkita epätyypillisissä yleistyneissä epileptikoissa.
Maen puuttuva heritabiliteetti. Jos geneettinen testaus on negatiivinen MAE yli 80% yksilöistä, missä on piilotettu geneettinen taakka? Vuoteen 2020 mennessä ehdottaisin kahta mahdollista selitystä. Ensinnäkin yleistyneiden epileptikkojen geneettinen taakka ei ole täysin tuntematon. Yli 30% väestöriskistä selittyy sellaisilla sairauksilla kuin lapsuusiän Poissaoloepilepsia (CAE) tai nuoruusiän myokloninen epilepsia (Jme). Selitystä eivät kuitenkaan anna yhden geenin syyt, vaan polygeeninen riski, viitaten tuhansien yhteisten geenivarianttien additiiviseen vaikutukseen. Voidaan olettaa, että MAE voi edustaa ”äärimmäistä fenotyyppiä” yleistyneistä epilepsioista, joissa tämä polygeeninen riski on erityisen suuri. Tämä hypoteesi on testattavissa ja sitä tullaan varmasti jatkamaan tulevaisuudessa. Lisäksi, on enemmän ihmisen genomi kuin exome-viimeaikaiset havainnot epileptikot ovat korostaneet roolia ei-koodaus variantteja, kuten toistaa laajennuksia familiaalinen aikuisten myokloninen epilepsia (FAME). Koska monet Mae: tä sairastavat lapset ovat hämmästyttävän samanlaisia kliinisiä ominaisuuksia, voi silti olla järkevää etsiä yhteistä geneettistä syytä monille Mae: tä sairastaville lapsille exomen ulkopuolella käyttäen koko genomin sekvensointia. Polygeeni riskipisteiden (PRS) ja whole genome sequencing (WGS) arviointi yhdessä ovat kaksi mahdollista keinoa selvittää mae: n geneettinen perusta.
mitä sinun tarvitsee tietää. Doosen oireyhtymä (MAE) on oletettavasti geneettinen epilepsiasyndrooma, jonka aiheuttaja on heterogeeninen. Tautia aiheuttavia variantteja esiintyy 14%: lla lapsista, ja ne tunnistetaan tyypillisesti lapsilla, joilla on muita neurologisia kehitysominaisuuksia, kuten kehitysviivettä tai autismia. Geenien triadi, mukaan lukien SYNGAP1, NEXMIF (KIAA2022) ja SLC6A1, ovat toistuvia geneettisiä syitä, jotka voivat viitata yhteiseen taustalla olevaan biologiaan, joka eroaa jonkin verran muista kehityshäiriöistä ja epileptisistä enkefalopatioista.

Ingo Helbig on lastenneurologi ja epilepsiagenetiikan tutkija, joka työskentelee Philadelphian lastensairaalassa (CHOP) Yhdysvalloissa. Hän johtaa myös epilepsiagenetiikan ryhmää Kielin yliopistossa Saksassa.

