MAE. Számos különböző gyermekkori epilepsziás szindróma létezik, amelyekről kritikusan tudatában vagyunk a genomikus korszakban, mivel kiemelkedő genetikai okokhoz kapcsolódnak, beleértve a Dravet-szindrómát (SCN1A) és a migráló fókuszos rohamokkal járó csecsemőkori epilepsziát (KCNT1). Számos más epilepsziás szindróma létezik, ahol genetikai ok már régóta gyanítható, de megfoghatatlan maradt. Az egyik epilepsziás szindróma, amely nagyrészt feltáratlan maradt, a Doose-szindróma, más néven Myoclonic Astatic epilepszia (MAE) vagy myoclonic-Atonic rohamokkal járó epilepszia. Egy nemrégiben végzett epilepsziás tanulmányban a Doose-szindróma genetikai architektúráját tártuk fel, és az egyének 14% – ánál azonosítottuk a monogénes okokat, beleértve a SYNGAP1-et, a NEXMIF-et (KIAA2022) és az SLC6A1-et. Tanulmányunk azt sugallja, hogy a Doose-szindróma genetikailag heterogén, esetleg különálló genetikai tájjal.
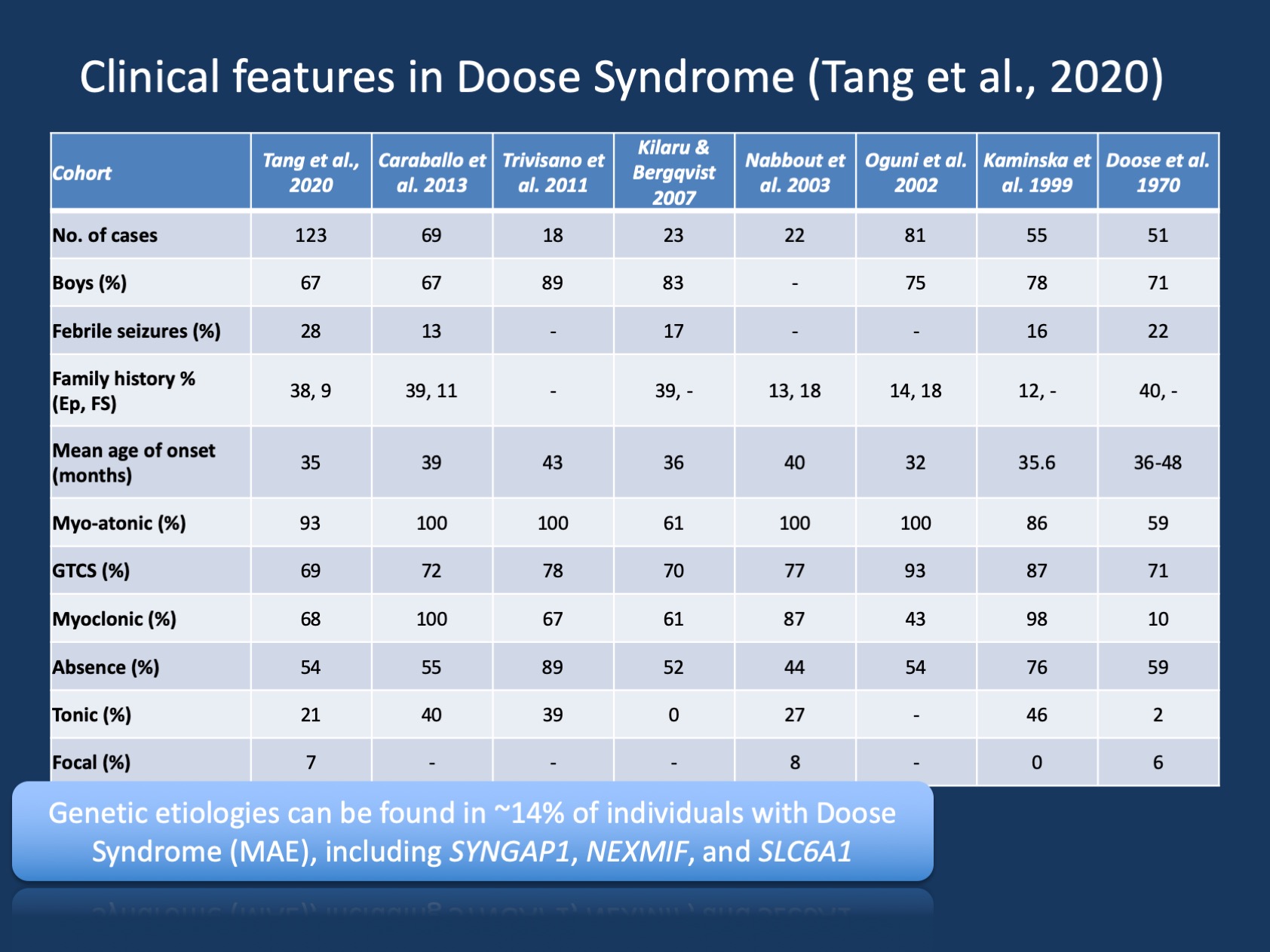
1. ábra. A Doose-szindróma klinikai jellemzői (epilepszia myoclonusos atonikus rohamokkal vagy myoclonusos asztatikus epilepsziával ). A táblázat a Tang et al., 2020-ban összehasonlítja a Doose-szindróma nagy kohorszainak klinikai jellemzőit, amelyekről az 1970-es kezdeti leírás óta számoltak be. A Doose-szindróma (MAE) ritka epilepsziás szindróma, de felismerhető klinikai entitás. Ellentétben azonban a Dravet-szindrómával, ahol az egyének >90%-ánál vannak betegséget okozó variánsok az SCN1A-ban, a Doose-szindróma genetikailag heterogén.
Mae története. Először is, hadd tisztázzak néhány névadási kérdést. A Doose-szindrómát vagy a Myoclonic Astatic epilepsziát (MAE) hivatalosan mioklonikus-atonikus rohamokkal járó epilepsziának nevezik a legfrissebb elnevezési konvenció alkalmazásával. Azonban, miközben általában megpróbáljuk összehangolni magunkat az epilepsziával kapcsolatos kifejezések használatának hivatalos módjával, hadd legyek itt egy kicsit makacs. A németországi Kieli Neuropediatrics Tanszéken képzett gyermekneurológusként ezt az állapotot Doose-szindrómának fogom nevezni, a Mae-t rövidítésként használva, Hermann Doose (1927-2018) tiszteletére, aki az Európai gyermekepileptológia egyik alapítója (link a ILAE In memoriam). Sok angol anyanyelvű általában rosszul ejti a kiejtést – a helyes kiejtés a “Dohs-ah”, és nem rímel a”liba” – ra.
a Mae klinikai képe. A Doose-nak általában fontos klinikai megfigyelést tulajdonítanak: az 1960-as években ő és munkatársai Kielben felismerték a gyermekek szokatlan epilepsziáját, amelyet hirtelen fellépő csepprohamok (atonikus rohamok), myoclonusos rohamok, generalizált tónusos-klónusos rohamok és általános EEG-minta jellemeztek, jellemzően két és öt éves kor között. Ez az epilepszia jellemzően olyan gyermekeknél fordult elő, akiknél korábban jellemző volt a fejlődés (lásd alább), és gyakran fejlődési késleltetést és értelmi fogyatékosságot eredményezett. Doose és munkatársai felismerték, hogy ez az újonnan elismert epilepsziás szindróma különbözik a Lennox-Gastaut-szindrómától vagy a láz által kiváltott generalizált tónusos-klónusos rohamokkal járó epilepsziáktól, amelyek közül az utóbbit később Dravet-szindrómaként fogalmazták meg.
megértése MAE. A Doose-szindróma (Mae) klinikai leírása az évek során állandó volt, de a Mae nem kapott annyi figyelmet, mint a gyermekkori epilepsziás szindrómák, ahol genetikai alapot fedeztek fel, és ahol a célzott kutatások lehetővé tehetik az új kezeléseket. Tang és munkatársai nemrégiben megjelent kiadványunkban nyolc tanulmányt soroltunk fel az elmúlt 50 évben 1970 és 2020 között, összesen 442 leírással. Ennek megfelelően a MAE ritka, de elég gyakori, hogy a legtöbb gyermek neurológus és gyermek epileptológus tisztában van ezzel a feltétellel. A MAE-ben szenvedő egyéneknek csak egy kis részénél vannak strukturális agyi eredmények vagy anyagcsere – rendellenességek, amelyek megmagyarázzák a mögöttes epilepsziát-a MAE-ben szenvedő gyermekek túlnyomó többségében az idegképalkotás és az anyagcsere-munka nem figyelemre méltó.
A MAE okának megtalálása. A magyarázó képalkotás vagy az anyagcsere-leletek hiánya aggodalomra ad okot egy mögöttes genetikai ok miatt. Míg a korábbi tanulmányok epidemiológiai vizsgálatokkal próbálták meghatározni a genetikai hozzájárulást a családtörténetek értékelésével, ezt a hagyományos módszert nagyrészt felváltották az epilepszia genetikájának legújabb felfedezései. A súlyos gyermekkori epilepsziák genetikai alapjának felmérése ma már a molekuláris genetika, nem pedig a genetikai epidemiológia révén történik. A Mae genetikai felépítésének értékelése volt az egyik cél Tang és munkatársai által az Epilepsia-ban közzétett tanulmányunkban.
A MAE egy betegség? Mielőtt leírnám a Mae genetikai eredményeit, hadd mutassak be egy folyamatban lévő vitát a MAE területén. A Doose és munkatársai által készített Első leírás nagyrészt olyan gyermekeket tartalmazott, akiknek tipikus fejlődése a roham kezdete előtt volt (>75%), ami összehasonlítható a legutóbbi tanulmányunkkal (79%). Ha azonban összehasonlítjuk a gyermekeket előzetes fejlődési problémákkal vagy anélkül, ugyanazt a betegséget vizsgáljuk, vagy csak a korábban figyelemre méltó fejlődésű gyermekeket kell tipikus Mae-nek tekinteni? Bár ez a kérdés nem könnyen megoldható, ha figyelembe vesszük az epilepszia történetét és kimenetelét, a mögöttes genetikai változások adhatnak néhány nyomot. A genetikai okokat gyakrabban azonosítják a súlyosabb neurodevelopmentális rendellenességekben szenvedő gyermekeknél, és minden azonosított genetikai okú gyermeknek az epilepszia mellett komplex neurodevelopmentális rendellenességei voltak. Ennek megfelelően genetikai okok figyelhetők meg MAE-ben szenvedő gyermekeknél, de jellemzően csak MAE-ben és komorbid neurodevelopmentális rendellenességekben szenvedő gyermekeknél, ami arra utal, hogy a Mae nemcsak genetikailag heterogén állapot, hanem genetikai heterogenitásuk és komorbiditásuk tekintetében hasonló tulajdonságokkal rendelkezik, mint más gyermekkori epilepszia. Például West-szindróma (infantilis görcsök) esetén a genetikai okokat csak neurodevelopmentális leletekkel rendelkező gyermekeknél azonosítják, nem pedig olyan gyermekeknél, akik közvetlenül reagálnak a gyógyszeres kezelésre neurodevelopmentális következmények nélkül.
genetikai leletek MAE-ben. A Mae genetikai spektruma más, mint általában az epilepsziás encephalopathiákban. A genetikai vizsgálatokat 85 egyénen végezték, és 12/85 (14%) egyénnek volt pozitív genetikai eredménye. Három gént találtak két személyben, köztük a SYNGAP1, a NEXMIF (KIAA2022) és az SLC6A1. A gének ezen hármasát korábban összefüggésbe hozták a generalizált rohamokkal járó fejlődési és epilepsziás encephalopathiákkal. Ezenkívül egyetlen egyednél találtak betegséget okozó variánsokat a KCNA2, az SCN2A, az STX1B, a KCNB1 és a MECP2-ben. Ezeket a géneket korábban a blogunkban tárgyaltuk, és ismert, hogy generalizált rohamokkal és generalizált EEG-jellemzőkkel rendelkeznek.
hiányzó gének a MAE-ben. Míg néhány korábbi jelentés variánsokat talált az SCN1A – ban, ezt nem találtuk kohorszunkban. Emellett más gyakori epilepsziás gének is hiányoznak, köztük a CDKL5 és az SLC2A1 (GLUT1). Ez azt sugallja, hogy a Mae genetikai tájképe némileg különbözik a többi fejlődési és epilepsziás encephalopathiától, annak ellenére, hogy viszonylag kis kohorszokból csak korlátozott következtetéseket lehet levonni. A Mae diagnosztikai hozama nem túl magas – 14% sokkal alacsonyabb, mint általában a fejlődési és epilepsziás encephalopathiákban. A tipikus fejlődésű egyének kizárásakor azonban a diagnosztikai hozamok sokkal magasabbak, ami arra utal, hogy a genetikai tesztelés értékes a Mae-ben szenvedő gyermekeknél. Általában, annak ellenére, hogy egyetlen Mae-ben szenvedő egyénnek sem volt betegséget okozó variánsa az SLC2A1-ben, a genetikai epilepsziák kezelhető okainak genetikai vizsgálatát általában atipikus generalizált epilepsziákban lehet figyelembe venni.
a Mae hiányzó örökölhetősége. Ha a genetikai teszt negatív A MAE-re az egyének több mint 80% – ában, hol van a rejtett genetikai teher? 2020-tól két lehetséges magyarázatot javasolnék. Először is, az Általános epilepsziák genetikai terhe nem teljesen ismeretlen. A lakosság több mint 30% – a kockázatot jelent olyan állapotokra, mint a gyermekkori hiány epilepszia (CAE) vagy a juvenilis myoclonusos epilepszia (JME). Ezt a magyarázatot azonban nem az egygénes okok adják, hanem a poligén kockázat, utalva több ezer közös genetikai variáns additív hatására. Feltételezhető, hogy a MAE az általánosított epilepsziák “extrém fenotípusát” jelentheti, ahol ez a poligén kockázat különösen magas. Ez a hipotézis tesztelhető, és a jövőben mindenképpen követni fogják. Ezenkívül az emberi genomban több van, mint az exome – az epilepsziák legújabb eredményei hangsúlyozták a nem kódoló variánsok szerepét, például a familiáris felnőtt myoclonusos epilepsziában (FAME). Tekintettel sok Mae-ben szenvedő gyermek feltűnően hasonló klinikai jellemzőire, még mindig ésszerű lehet közös genetikai okot keresni sok Mae-vel rendelkező gyermek számára az exómán kívül, teljes genomszekvenálás alkalmazásával. Összességében a poligén kockázati pontszámok (PRS) és a teljes genom szekvenálás (WGS) értékelése két lehetséges út a Mae genetikai alapjának további azonosítására.
mit kell tudni. A Doose-szindróma (MAE) Feltehetően genetikai epilepsziás szindróma, heterogén okokkal. A betegséget okozó variánsok a gyermekek 14% – ában vannak jelen, és jellemzően olyan gyermekeknél azonosíthatók, akiknek további neurodevelopmentális jellemzői vannak, mint például a fejlődési késleltetés vagy az autizmus. A gének hármasa, köztük a SYNGAP1, a NEXMIF (KIAA2022) és az SLC6A1 visszatérő genetikai okok, amelyek utalhatnak egy közös mögöttes biológiára, amely némileg különbözik a többi fejlődési és epilepsziás encephalopathiától.

Ingo Helbig egy gyermek neurológus és epilepszia genetika kutató dolgozik a Children ‘ s Hospital of Philadelphia (CHOP), USA. Az epilepszia genetikai csoportját is vezeti a németországi Kieli Egyetemen.

