MAE. Det er mange forskjellige barndomsepilepsi syndromer som vi har blitt kritisk oppmerksomme på i den genomiske epoken, da de er knyttet til fremtredende genetiske årsaker, inkludert Dravet Syndrom (SCN1A) Og Epilepsi Av Barndom med Migrerende Fokale Anfall (KCNT1). Det er imidlertid mange andre epilepsi syndromer der en genetisk årsak har lenge vært mistenkt, men har vært unnvikende. En av epilepsi syndromer som i stor grad har forblitt uutforsket Er Doose Syndrom, også referert Til Som Myoklon Astatisk Epilepsi (MAE) Eller Epilepsi Med Myoklon – Atoniske Anfall. I En nylig Studie i Epilepsi utforsket vi Den genetiske arkitekturen Til Doosesyndrom og identifiserte monogene årsaker hos 14% av individer, inkludert SYNGAP1, NEXMIF (KIAA2022) og SLC6A1. Vår studie tyder på At Doosesyndrom er genetisk heterogent, muligens med et tydelig genetisk landskap.
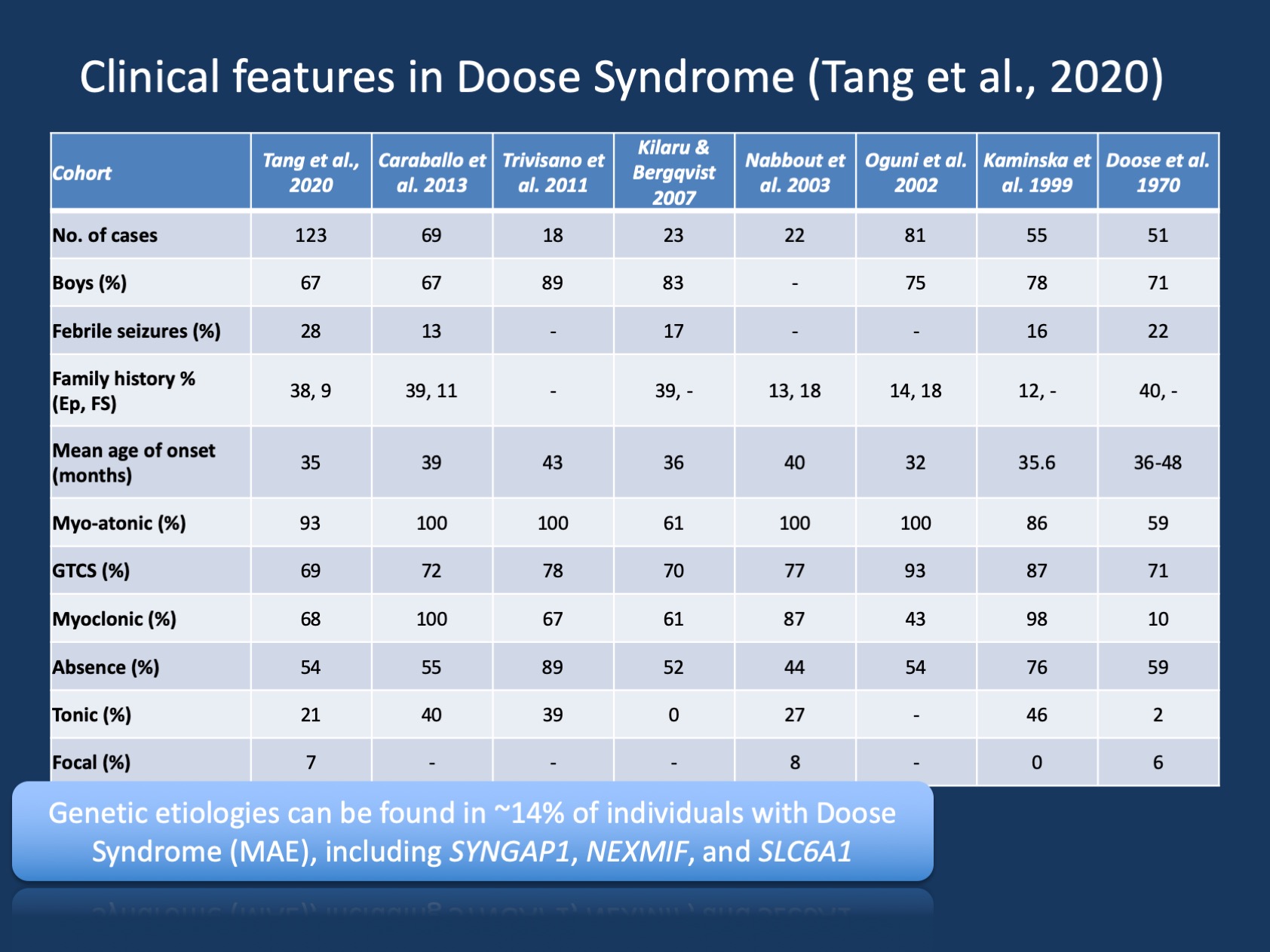
Figur 1. Kliniske trekk Ved Doosesyndrom(epilepsi med myoklone atoniske anfall eller Myoklon Astatisk Epilepsi). Tabellen er tilpasset fra vår siste publikasjon Av Tang et al., 2020 og sammenligner de kliniske egenskapene i de store kohortene Av Doose Syndrom som har blitt rapportert siden den første beskrivelsen i 1970. Doose Syndrom (MAE) er en sjelden epilepsi syndrom, men er en gjenkjennelig klinisk enhet. I motsetning Til Dravet Syndrom hvor >90% av individene har sykdomsfremkallende varianter I SCN1A, Er Doosesyndrom genetisk heterogent.
HISTORIEN OM MAE. Først av alt, la meg rydde opp noen navngivningsproblemer. Doose Syndrom Eller Myoklon Astatisk Epilepsi (MAE) er nå offisielt referert Til Som Epilepsi Med Myoklon – Atoniske Anfall ved hjelp av den nyeste navnekonvensjonen. Men mens vi vanligvis prøver å justere oss med den offisielle måten å bruke epilepsi – relaterte termer på, la meg være litt sta her. Som barn nevrolog utdannet Ved Institutt For Neuropediatrics I Kiel, Tyskland, vil jeg referere til denne tilstanden Som Doose Syndrom ved HJELP AV MAE som en stenografi, hedre Hermann Doose (1927-2018) som er en av grunnleggerne Av Europeisk pediatrisk epileptologi (link TIL ILAE in memoriam). Mange engelsktalende får vanligvis uttalen feil – den riktige uttalen er «Dohs-ah» og rimer IKKE med «gås».
det kliniske bildet AV MAE. Doose er vanligvis kreditert med å gjøre en viktig klinisk observasjon: på 1960-tallet anerkjente han og hans samarbeidspartnere I Kiel en uvanlig epilepsi hos barn som var preget av plutselig utbrudd av dråpeangrep (atoniske anfall), myokloniske anfall, generaliserte tonisk-kloniske anfall og et generalisert EEG-mønster, som vanligvis forekommer mellom to og fem år. Denne epilepsien oppstod vanligvis hos barn med tidligere typisk utvikling (se nedenfor) og resulterte ofte i utviklingsforsinkelse og intellektuell funksjonshemning. Doose og samarbeidspartnere anerkjente at dette nylig anerkjente epilepsisyndromet var forskjellig fra Lennox-Gastaut Syndrom eller epilepsier med generaliserte tonisk-kloniske anfall utfelt av feber, sistnevnte ble senere konseptualisert Som Dravet Syndrom.
Forstå MAE. Kliniske beskrivelser Av Doose Syndrom (MAE) har vært jevn gjennom årene, MEN MAE har ikke fått så mye oppmerksomhet som barndoms epilepsi syndromer hvor en genetisk basis hadde blitt oppdaget og hvor målrettet forskning kan tillate nye behandlinger. I Vår siste publikasjon Av Tang og samarbeidspartnere har Vi listet opp åtte studier de siste 50 årene mellom 1970 og 2020 med totalt 442 beskrevne individer. Følgelig ER MAE sjelden, men det er vanlig nok at de fleste barnenevrologer og pediatriske epileptologer er klar over denne tilstanden. Bare en liten delmengde av personer med MAE har strukturelle hjernefunn eller metabolske abnormiteter som forklarer den underliggende epilepsien – hos de aller fleste barn med MAE er neuroimaging og metabolsk opparbeidelse unremarkable.
Finne årsaken TIL MAE. Fraværet av forklarende bildebehandling eller metabolske funn øker bekymringen for en underliggende genetisk årsak. Mens tidligere studier hadde forsøkt å peke på et genetisk bidrag gjennom epidemiologiske studier ved å vurdere familiehistorier, har denne tradisjonelle metoden i stor grad blitt erstattet av nyere funn i epilepsi genetikk. Å vurdere det genetiske grunnlaget for alvorlige barndomsepilepsi er nå gjort gjennom molekylær genetikk i stedet for genetisk epidemiologi. Vurdering av maes genetiske arkitektur var et av målene I Vår studie Av Tang og samarbeidspartnere som ble publisert I Epilepsia.
ER MAE en sykdom? Før jeg beskriver de genetiske funnene I MAE, la MEG introdusere en pågående debatt innen MAE-feltet. Den første beskrivelsen av Doose og samarbeidspartnere inkluderte i stor grad barn med typisk utvikling før anfallsutbrudd (>75%), som er sammenlignbar med vår nylige studie (79%). Men når vi sammenligner barn med og uten tidligere utviklingsproblemer, ser vi på samme sykdom eller bør bare barn med tidligere unremarkable utvikling betraktes som typiske MAE? Selv om dette spørsmålet ikke er lett løst når man vurderer epilepsihistorier og utfall, kan de underliggende genetiske endringene gi noen ledetråder. Genetiske årsaker identifiseres oftere hos barn med mer alvorlige nevroutviklingsforstyrrelser, og alle barn med identifiserte genetiske årsaker hadde komplekse nevroutviklingsforstyrrelser i tillegg til epilepsi. Følgelig kan genetiske årsaker observeres hos BARN MED MAE, men vanligvis bare hos barn med MAE og komorbide nevroutviklingsforstyrrelser, noe som tyder PÅ AT MAE ikke bare er en genetisk heterogen tilstand, men har lignende egenskaper som annen epilepsi i barndommen med hensyn til deres genetiske heterogenitet og komorbiditet. For Eksempel, I West Syndrom (Infantile Spasmer), er genetiske årsaker også bare identifisert hos barn med nevrodevelopmental funn og ikke hos barn som direkte reagerer på medisinering uten nevrodevelopmental følger.
Genetiske funn I MAE. Det genetiske spekteret I MAE er annerledes enn i epileptiske encefalopatier som helhet. Genetisk testing ble utført hos 85 individer og 12/85 (14%) individer hadde positive genetiske funn. Tre gener ble funnet i to individer, inkludert SYNGAP1, NEXMIF (KIAA2022) OG SLC6A1. Denne triaden av gener har tidligere vært knyttet til utviklings-og epileptiske encefalopatier med generaliserte anfall. I tillegg ble enkeltpersoner funnet med sykdomsfremkallende varianter I KCNA2, SCN2A, STX1B, KCNB1 og MECP2. Alle disse genene ble tidligere diskutert på bloggen vår og er kjent for å presentere med generaliserte anfall og generaliserte EEG-funksjoner.
Fraværende gener I MAE. Mens noen tidligere rapporter har funnet varianter I SCN1A, ble dette ikke funnet i vår kohort. I tillegg er andre vanlige epilepsi gener spesielt fraværende, inkludert CDKL5 OG SLC2A1 (GLUT1). Dette antyder at det genetiske landskapet TIL MAE er noe distinkt og forskjellig fra andre utviklings-og epileptiske encefalopatier, selv om bare begrensede konklusjoner kan trekkes fra relativt små kohorter. Diagnostisk utbytte I MAE er ikke veldig høyt-14% er mye lavere enn vanligvis sett i utviklings-og epileptiske encefalopatier. Men når man utelukker personer med typisk utvikling, er de diagnostiske utbyttene mye høyere, noe som tyder på at genetisk testing er av verdi hos barn med MAE. Generelt, selv om ingen enkeltperson med MAE hadde sykdomsfremkallende varianter I SLC2A1, kan genetisk testing for behandlingsårsaker til genetiske epilepsier generelt vurderes ved atypiske generaliserte epilepsier.
den manglende arvbarheten TIL MAE. Hvis genetisk testing er negativ FOR MAE hos mer enn 80% av individer, hvor er den skjulte genetiske byrden? Fra 2020 vil jeg foreslå to mulige forklaringer. For det første er den genetiske byrden av generaliserte epilepsier ikke helt ukjent. Mer enn 30% av populasjonsrisikoen for tilstander som Barndomsfraværsepilepsi (CAE) eller Juvenil Myoklon Epilepsi (JME) er forklart. Denne forklaringen er imidlertid ikke gitt av enkeltgenårsaker, men av polygenisk risiko, med henvisning til additiv effekt av tusenvis av vanlige genetiske varianter. DET kan antas at MAE kan representere en «ekstrem fenotype» av generaliserte epilepsier der denne polygeniske risikoen er spesielt høy. Denne hypotesen er testbar og vil definitivt bli forfulgt i fremtiden. I tillegg er det mer til det menneskelige genomet enn exome – nylige funn i epilepsiene har lagt vekt på rollen som ikke-kodende varianter, for eksempel gjentatte utvidelser I Familiær Voksen Myoklonisk Epilepsi (FAME). Gitt de slående lignende kliniske egenskapene hos mange barn MED MAE, kan det fortsatt være rimelig å se etter en felles genetisk årsak for mange barn med MAE utenfor exome, ved hjelp av hele genomsekvensering. Ved å ta sammen, vurdere polygenic risk score (PRS) og wgs (whole genome sequencing) er to potensielle veier for å identifisere det genetiske grunnlaget for MAE.
Hva du trenger å vite. Doose Syndrom (MAE) er et antagelig genetisk epilepsi syndrom med en heterogen årsak. Sykdomsfremkallende varianter er tilstede hos 14% av barna og er vanligvis identifisert hos barn med ekstra nevroutviklingsfunksjoner som utviklingsforsinkelse eller autisme. En triade av gener, INKLUDERT SYNGAP1, NEXMIF (KIAA2022) og SLC6A1, er tilbakevendende genetiske årsaker som kan antyde en felles underliggende biologi som er noe forskjellig fra andre utviklings-og epileptiske encefalopatier.

Ingo Helbig er barnenevrolog og forsker på epilepsi-genetikk ved Children ‘ S Hospital Of Philadelphia (CHOP), USA. Han leder også gruppen epilepsi genetikk ved Universitetet I Kiel, Tyskland.

