MAE. Er zijn veel verschillende epilepsiesyndromen bij kinderen waarvan we ons in het genomische Tijdperk kritisch bewust zijn geworden omdat ze verbonden zijn met prominente genetische oorzaken, waaronder het Dravet-syndroom (SCN1A) en epilepsie van de kindertijd met migrerende focale aanvallen (KCNT1). Nochtans, zijn er vele andere epilepsiesyndromen waar een genetische oorzaak al lang wordt vermoed, maar ongrijpbaar is gebleven. Een van de epilepsiesyndromen die grotendeels onontgonnen is gebleven is syndroom Doose, ook aangeduid als Myoclonic Astatic epilepsie (MAE) of epilepsie met Myoclonic-Atonic aanvallen. In een recente studie bij epilepsie onderzochten we de genetische architectuur van het Doose syndroom en identificeerden we monogene oorzaken bij 14% van de individuen, waaronder SYNGAP1, NEXMIF (KIAA2022) en SLC6A1. Onze studie suggereert dat Doose syndroom genetisch heterogeen is, mogelijk met een duidelijk genetisch landschap.
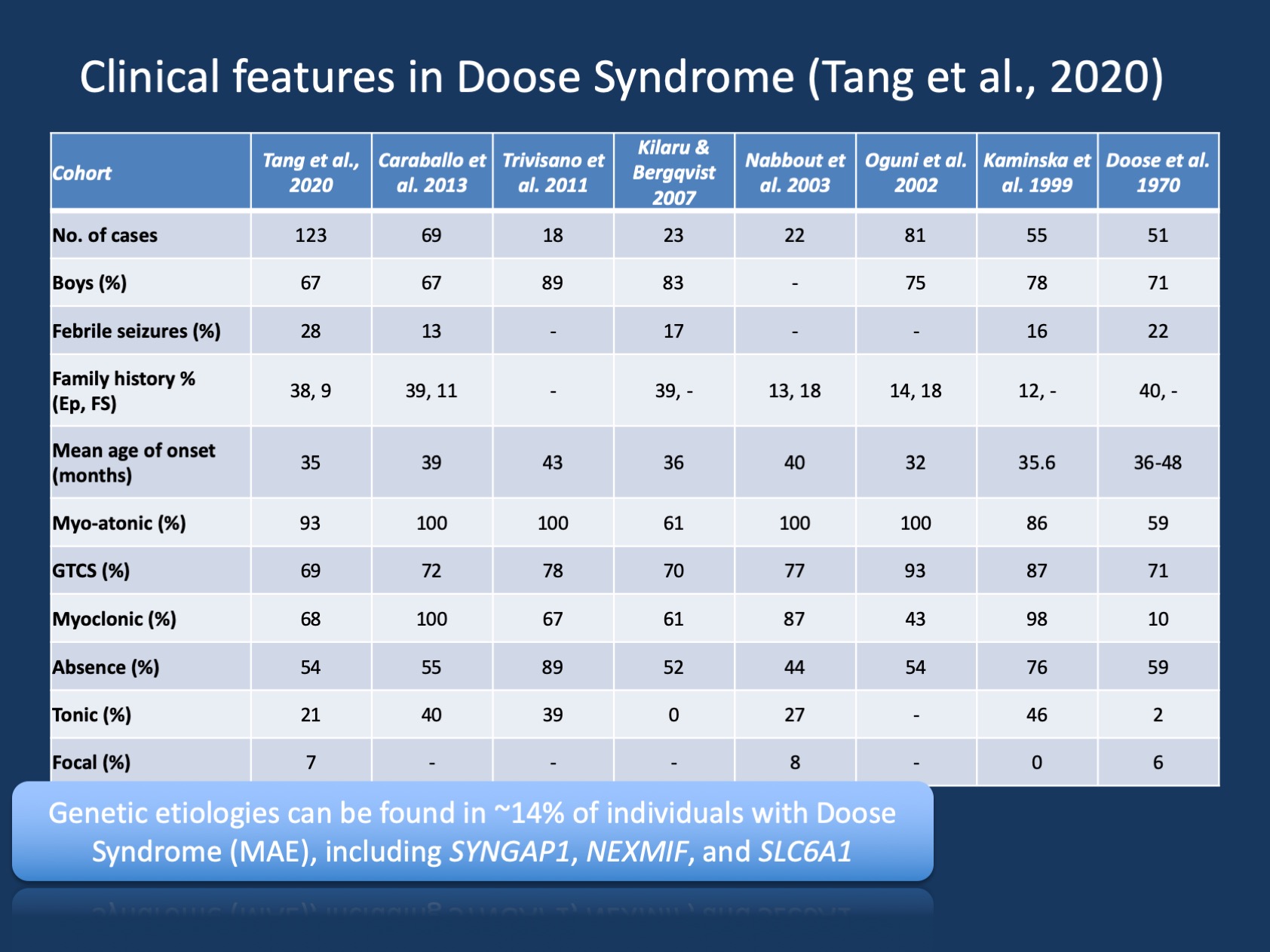
figuur 1. De klinische kenmerken van het Doose-syndroom (epilepsie met myoklonische atonische aanvallen of myoklonische astatische epilepsie ). De tabel is aangepast van onze recente publicatie door Tang et al., 2020 en vergelijkt de klinische eigenschappen in de grote cohorten van Doose syndroom die sinds de aanvankelijke beschrijving in 1970 zijn gemeld. Doose syndroom (MAE) is een zeldzame epilepsie syndroom, maar is een herkenbare klinische entiteit. Echter, in tegenstelling tot het syndroom van Dravet, waar >90% van de personen ziekteveroorzakende varianten in SCN1A hebben, is het syndroom van Doose genetisch heterogeen.
de geschiedenis van MAE. Allereerst wil ik wat naamgevingsproblemen ophelderen. Doose syndroom of Myoclonic Astatic epilepsie (MAE) wordt nu officieel aangeduid als epilepsie met Myoclonic-Atonic aanvallen met behulp van de meest recente naamgevingsconventie. Hoewel we meestal proberen om ons aan te sluiten bij de officiële manier van hoe epilepsiegerelateerde termen worden gebruikt, laat ik hier een beetje koppig zijn. Als kinderneuroloog opgeleid aan de afdeling Neuropediatrie in Kiel, Duitsland, zal ik deze aandoening noemen als Doose syndroom met behulp van MAE als steno, ter ere van Hermann Doose (1927-2018) die een van de grondleggers is van de Europese pediatrische Epileptologie (link naar ilae In memoriam). Veel Engels native speakers meestal krijgen de uitspraak verkeerd – de juiste uitspraak is ” Dohs-ah “en rijmt niet met”gans”.
het klinische beeld van MAE. Doose wordt meestal gecrediteerd met het maken van een belangrijke klinische observatie: in de jaren 60 onderkenden hij en zijn medewerkers in Kiel een ongebruikelijke epilepsie bij kinderen die werd gekenmerkt door plotselinge aanvallen van vallen (atonische aanvallen), myoklonische aanvallen, gegeneraliseerde tonisch-clonische aanvallen en een gegeneraliseerd EEG-patroon, meestal tussen de leeftijd van twee en vijf. Deze epilepsie kwam typisch voor bij kinderen met voorafgaande typische ontwikkeling (zie hieronder) en resulteerde vaak in ontwikkelingsachterstand en intellectuele handicap. Doose en medewerkers erkenden dat dit nieuw erkende epilepsiesyndroom anders was dan het syndroom van Lennox-Gastaut of epilepsie met gegeneraliseerde tonisch-clonische aanvallen die werden neergeslagen door koorts, waarvan de laatste later werd geconceptualiseerd als het syndroom van Dravet.
inzicht MAE. De klinische beschrijvingen van Doose syndroom (MAE) zijn regelmatig in de loop van de jaren geweest, maar MAE heeft niet zo veel aandacht zoals de syndromen van de kindertijd epilepsie ontvangen waar een genetische basis was ontdekt en waar het gerichte onderzoek voor nieuwe behandelingen kan toestaan. In onze recente publicatie van Tang and collaborators hebben we acht studies in de afgelopen 50 jaar tussen 1970 en 2020 opgesomd met in totaal 442 beschreven individuen. Dienovereenkomstig is MAE zeldzaam, maar het is algemeen genoeg dat de meeste kindneurologen en pediatrische epileptologen zich bewust zijn van deze aandoening. Slechts een kleine subset van individuen met MAE heeft structurele hersenenbevindingen of metabolische abnormaliteiten die de onderliggende epilepsie verklaren-in de overgrote meerderheid van kinderen met MAE, is neuroimaging en metabolische work-up onopvallend.
de oorzaak van MAE vinden. De afwezigheid van verklarende weergave of metabolische bevindingen leidt tot de zorg voor een onderliggende genetische oorzaak. Terwijl de voorafgaande studies op een genetische bijdrage door epidemiologische studies hadden geprobeerd vast te spelden door familiegeschiedenis te beoordelen, is deze traditionele methode grotendeels door recente ontdekkingen in epilepsiegenetica vervangen. Het beoordelen van de genetische basis van ernstige epilepsies bij kinderen wordt nu gedaan door middel van Moleculaire Genetica in plaats van genetische epidemiologie. Het beoordelen van de genetische architectuur van MAE was een van de doelen in onze studie door Tang en medewerkers die werd gepubliceerd in Epilepsia.
Is één ziekte? Voorafgaand aan het beschrijven van de genetische bevindingen in MAE, laat me één lopend debat binnen het Mae-gebied introduceren. De initiële beschrijving door Doose en collaborateurs omvatte grotendeels kinderen met een typische ontwikkeling voorafgaand aan het begin van de aanval (>75%), wat vergelijkbaar is met onze recente studie (79%). Nochtans, wanneer het vergelijken van kinderen met en zonder voorafgaande ontwikkelingskwesties, kijken wij naar dezelfde ziekte of zouden alleen kinderen met voorafgaande onopvallende ontwikkeling als typische MAE moeten worden beschouwd? Terwijl deze vraag niet gemakkelijk wordt opgelost wanneer het overwegen van de epilepsiegeschiedenissen en resultaten, kunnen de onderliggende genetische veranderingen sommige aanwijzingen verstrekken. Genetische oorzaken worden vaker geà dentificeerd bij kinderen met ernstigere neurologische ontwikkelingsstoornissen en alle kinderen met geà dentificeerde genetische oorzaken hadden naast de epilepsie complexe neurologische ontwikkelingsstoornissen. Dienovereenkomstig, kunnen de genetische oorzaken in kinderen met MAE, maar typisch slechts in kinderen met MAE en comorbid neurodevelopmental wanorde worden waargenomen, die suggereren dat MAE niet alleen een genetisch heterogene voorwaarde is, maar gelijkaardige eigenschappen als andere epilepsie in kinderjaren met betrekking tot hun genetische heterogeniteit en comorbiditeit heeft. Bijvoorbeeld, bij het West-syndroom (infantiele spasmen) worden genetische oorzaken ook alleen geïdentificeerd bij kinderen met neurologische ontwikkelingsbevindingen en niet bij kinderen die direct reageren op medicatie zonder neurologische gevolgen.
genetische bevindingen in MAE. Het genetische spectrum in MAE is anders dan in de epileptische encefalopathieën in het algemeen. Genetische tests werden uitgevoerd bij 85 personen en 12/85 (14%) personen hadden positieve genetische bevindingen. Drie genen werden gevonden in twee individuen, met inbegrip van SYNGAP1, NEXMIF (KIAA2022), en SLC6A1. Deze triade van genen is eerder in verband gebracht met ontwikkelings – en epileptische encefalopathieën met gegeneraliseerde aanvallen. Daarnaast werden enkele individuen gevonden met ziekteveroorzakende varianten in KCNA2, SCN2A, STX1B, KCNB1 en MECP2. Al deze genen werden eerder besproken op onze blog en zijn bekend om te presenteren met gegeneraliseerde aanvallen en gegeneraliseerde EEG-functies.
afwezige genen in MAE. Hoewel sommige eerdere rapporten varianten in SCN1A hebben gevonden, werd dit niet gevonden in onze cohort. Bovendien zijn andere gemeenschappelijke epilepsiegenen met name afwezig, met inbegrip van CDKL5 en SLC2A1 (GLUT1). Dit stelt voor dat het genetische landschap van MAE enigszins verschillend en verschillend van andere ontwikkelings en epileptische encefalopathieën is, hoewel slechts beperkte conclusies uit vrij kleine cohorten kunnen worden getrokken. De kenmerkende opbrengst in MAE is niet zeer hoog-14% is veel lager dan typisch gezien in de ontwikkelings en epileptische encefalopathieën. Nochtans, wanneer het uitsluiten van individuen met typische ontwikkeling, is de kenmerkende opbrengst veel hoger, die voorstelt dat het genetische testen van waarde in kinderen met MAE is. In het algemeen, hoewel geen enkel individu met MAE ziekte-veroorzakende varianten in SLC2A1 had, kan het genetische testen voor behandelbare oorzaken van genetische epilepsies over het algemeen in atypische algemene epilepsies worden overwogen.
de ontbrekende erfelijkheid van MAE. Als het genetische testen voor MAE in meer dan 80% van individuen negatief is, waar is de verborgen genetische Last? Vanaf 2020 stel ik twee mogelijke verklaringen voor. Ten eerste is de genetische belasting van gegeneraliseerde epilepsie niet geheel onbekend. Meer dan 30% van het populatierisico voor aandoeningen zoals Kinderepilepsie (CAE) of juveniele myoclonische epilepsie (JME) wordt verklaard. Echter, deze verklaring wordt niet gegeven door single-gen oorzaken, maar door polygene risico, verwijzend naar het additieve effect van duizenden gemeenschappelijke genetische varianten. Men zou kunnen veronderstellen dat MAE een “extreem fenotype” van gegeneraliseerde epilepsie kan vertegenwoordigen waar dit polygene risico bijzonder hoog is. Deze hypothese is toetsbaar en zal zeker in de toekomst worden nagestreefd. Daarnaast is er meer aan het menselijk genoom dan de exome – recente bevindingen in de epilepsies hebben de rol van niet-coderende varianten benadrukt, zoals herhaalde uitbreidingen in familiale volwassen myoclonische epilepsie (FAME). Gezien de opvallend gelijkaardige klinische eigenschappen in vele kinderen met MAE, kan het nog redelijk zijn om een gemeenschappelijke genetische oorzaak voor vele kinderen met MAE buiten exome te zoeken, gebruikend het gehele genoom rangschikken. Het samen nemen, het beoordelen van polygenic risicoscores (PRS) en het gehele genoom rangschikken (WGS) zijn twee potentiële wegen om de genetische basis van MAE verder te identificeren.
wat u moet weten. Doose syndroom (MAE) is een vermoedelijk genetisch epilepsiesyndroom met een heterogene oorzaak. Ziekte-veroorzakende varianten zijn aanwezig in 14% van kinderen en worden typisch geà dentificeerd in kinderen met extra neurodevelopmental eigenschappen zoals ontwikkelingsachterstand of autisme. Een triade van genen, waaronder SYNGAP1, NEXMIF (KIAA2022) en SLC6A1, zijn terugkerende genetische oorzaken die kunnen wijzen op een gedeelde onderliggende biologie die enigszins verschilt van andere ontwikkelings-en epileptische encefalopathieën.

Ingo Helbig is een kinderneuroloog en epilepsie genetica onderzoeker werkzaam bij het Children ‘ s Hospital van Philadelphia (CHOP), USA. Hij leidt ook de groep epilepsie genetica aan de Universiteit van Kiel, Duitsland.

