MAE. Istnieje wiele różnych dziecięcych zespołów padaczkowych, o których krytycznie zdajemy sobie sprawę w erze genomowej, ponieważ są one związane z wybitnymi przyczynami genetycznymi, w tym zespołem Draveta (SCN1A) i padaczką niemowlęctwa z migrującymi napadami ogniskowymi (Kcnt1). Istnieje jednak wiele innych zespołów padaczkowych, w których przyczyna genetyczna od dawna była podejrzewana, ale pozostała nieuchwytna. Jednym z zespołów padaczkowych, który w dużej mierze pozostał niezbadany, jest zespół Doose ’ a, zwany także miokloniczną padaczką Astatyczną (MAE) lub padaczką z napadami Miokloniczno-atonicznymi. W niedawnym badaniu w epilepsji zbadaliśmy architekturę genetyczną zespołu Doose ’ a i zidentyfikowaliśmy przyczyny monogenne u 14% osób, w tym SYNGAP1, NEXMIF (KIAA2022) i SLC6A1. Nasze badania sugerują, że zespół Doose ’ a jest genetycznie heterogeniczny, prawdopodobnie z wyraźnym krajobrazem genetycznym.
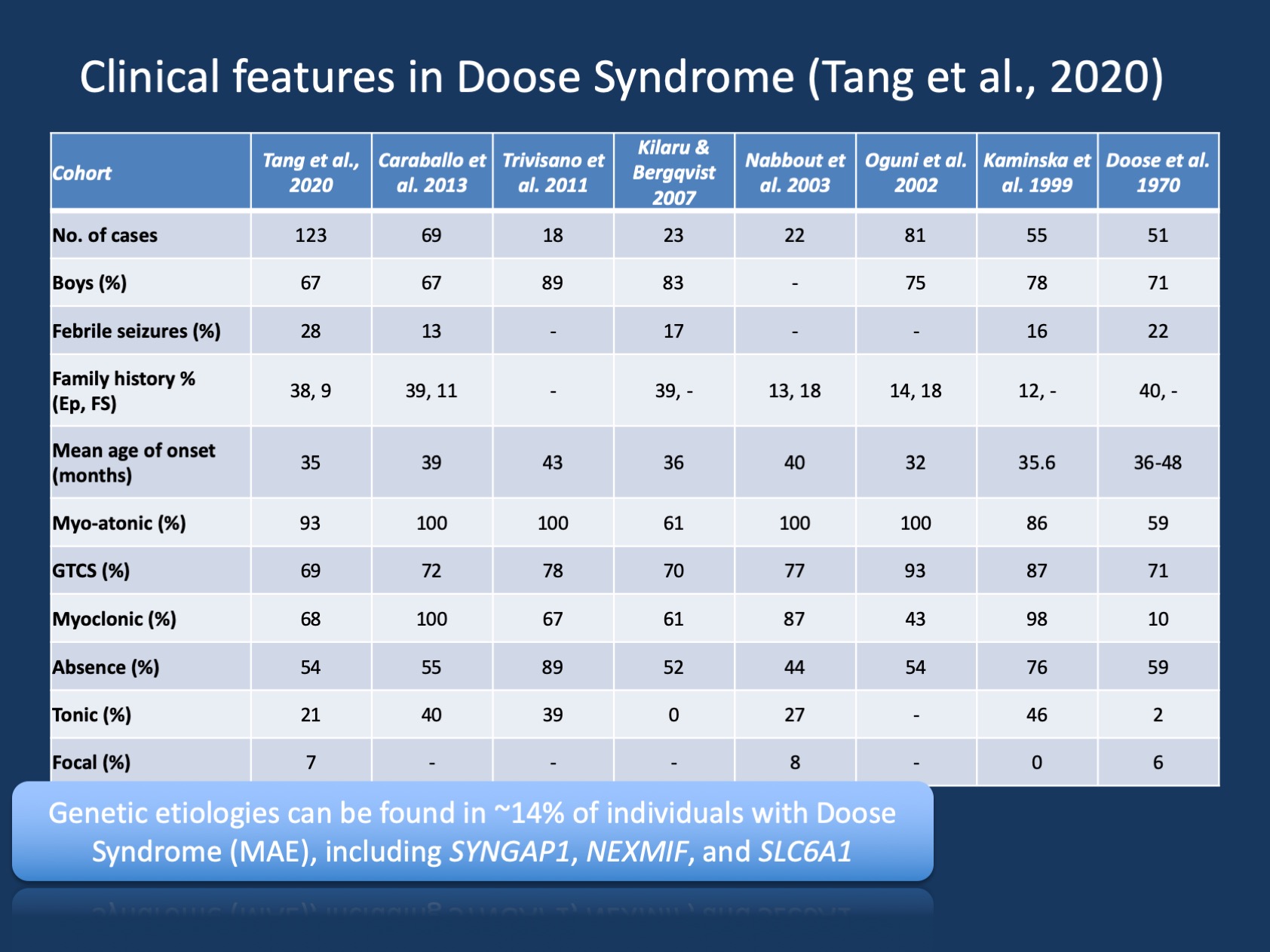
Rysunek 1. Cechy kliniczne zespołu Doose 'a (padaczka z napadami mioklonicznymi lub miokloniczną padaczką Astatyczną ). Tabela została zaadaptowana z naszej najnowszej publikacji przez Tang et al., 2020 i porównuje cechy kliniczne w dużych kohortach zespołu Doose ’ a, które odnotowano od początkowego opisu w 1970. Zespół doose ’ a (MAE) jest rzadkim zespołem padaczkowym, ale jest rozpoznawalnym podmiotem klinicznym. Jednak w przeciwieństwie do zespołu Dravet, gdzie >90% osób ma warianty chorobotwórcze w SCN1A, zespół Doose ’ a jest genetycznie heterogeniczny.
historia MAE. Po pierwsze, wyjaśnię kilka kwestii nazewnictwa. Zespół Doose ’ a lub miokloniczna epilepsja astatyczna (MAE) jest obecnie oficjalnie określana jako padaczka z napadami Miokloniczno-atonicznymi przy użyciu najnowszej konwencji nazewnictwa. Chociaż zazwyczaj staramy się dostosować do oficjalnego sposobu używania terminów związanych z epilepsją, pozwólcie, że będę nieco uparty. Jako neurolog dziecięcy przeszkolony w klinice Neuropediatrii w Kilonii, w Niemczech, będę nazywał ten stan syndromem Doose 'a używając skrótu MAE, honorując Hermanna Doose’ a (1927-2018), który jest jednym z założycieli Europejskiej Epileptologii dziecięcej (link do ILAE in memoriam). Wielu rodzimych użytkowników języka angielskiego zazwyczaj źle wymawia-poprawna wymowa to ” dohs-ah „i nie rymuje się z”goose”.
obraz kliniczny MAE. Doose ’ owi przypisuje się zwykle dokonanie ważnej obserwacji klinicznej: w latach 60.wraz ze współpracownikami w Kilonii rozpoznał nietypową padaczkę u dzieci, która charakteryzowała się nagłym wystąpieniem napadów kropli (napadów atonicznych), napadów mioklonicznych, uogólnionych napadów toniczno-klonicznych i uogólnionego wzorca EEG, Zwykle występującego w wieku od dwóch do pięciu lat. Padaczka ta występowała zazwyczaj u dzieci z wcześniejszym typowym rozwojem (patrz poniżej) i często powodowała opóźnienie rozwoju i niepełnosprawność intelektualną. Doose i współpracownicy uznali, że ten nowo rozpoznany zespół epilepsji różni się od zespołu Lennoxa-Gastauta lub epilepsji z uogólnionymi napadami toniczno-klonicznymi wytrąconymi przez gorączkę, z których ostatnie zostały następnie uznane za zespół Draveta.
Kliniczne opisy syndromu Doose ’ a (MAE) byly stabilne przez lata, ale MAE nie otrzymal tyle uwagi, co syndromy padaczki u dzieci, gdzie genetyczna podstawa zostala odkryta i gdzie ukierunkowane badania moga pozwolic na nowatorskie metody leczenia. W naszej najnowszej publikacji autorstwa Tang i współpracowników wymieniliśmy osiem badań w ciągu ostatnich 50 lat między 1970 a 2020 rokiem z łącznie 442 opisanymi osobami. W związku z tym MAE jest rzadkie, ale jest na tyle powszechne, że większość neurologów dziecięcych i epileptologów dziecięcych jest świadoma tego stanu. Tylko niewielki podzbiór osobniczy z MAE mieć strukturalny mózg odkrycie lub metaboliczny nieprawidłowość który wyjaśniać podstawowy padaczka – w zdecydowany większość dzieci z MAE, neuroobrazowanie i metaboliczny praca-up być nijaki.
Brak wyjaśniania obrazowania lub metabolicznych ustaleń budzi obawy o podstawowej przyczyny genetycznej. Podczas gdy wcześniejsze badania próbowały przypiąć wkład genetyczny poprzez badania epidemiologiczne poprzez ocenę historii rodziny, ta tradycyjna metoda została w dużej mierze zastąpiona przez ostatnie odkrycia w genetyce padaczki. Ocena genetycznych podstaw ciężkiej epilepsji u dzieci odbywa się obecnie za pomocą genetyki molekularnej, a nie epidemiologii genetycznej. Ocena architektury genetycznej MAE był jednym z celów w naszym badaniu przez Tang i współpracowników, który został opublikowany w Epilepsia.
czy MAE jest jedną chorobą? Przed opisaniem genetycznych znalezisk w MAE, pozwól mi przedstawic jeden trwajacy debata w ramach MAE dziedzinie. Wstępny opis doose i współpracowników w dużej mierze obejmował dzieci z typowym rozwojem przed wystąpieniem napadów (>75%), co jest porównywalne z naszym ostatnim badaniem (79%). Jednak porównując dzieci z wcześniejszymi problemami rozwojowymi i bez nich, czy patrzymy na tę samą chorobę, czy tylko dzieci z wcześniejszym, nijakim rozwojem powinny być uważane za typowe? Chociaż to pytanie nie jest łatwo rozwiązać, biorąc pod uwagę historię i wyniki padaczki, podstawowe zmiany genetyczne mogą dostarczyć pewnych wskazówek. Przyczyny genetyczne są identyfikowane częściej u dzieci z cięższymi zaburzeniami neurorozwojowymi, a wszystkie dzieci z zidentyfikowanymi przyczynami genetycznymi miały złożone zaburzenia neurorozwojowe oprócz padaczki. W związku z tym, genetyczne przyczyny mogą obserwować w dzieciach z MAE, ale typowo tylko w dzieciach z Mae i comorbid neurodevelopmental nieład, sugerując że Mae jest nie tylko genetycznie heterogeniczny warunek, ale ma podobne cechy jak inne epilepsja w dzieciństwie w odniesieniu do ich genetycznej heterogeniczności i comorbidity. Na przykład w zespole Westa (skurcze dziecięce) przyczyny genetyczne są również identyfikowane tylko u dzieci z odkryciami neurorozwojowymi, a nie u dzieci, które bezpośrednio reagują na leki bez następstw neurorozwojowych.
Spektrum genetyczne w MAE jest INNE niż w EPILEPTYCZNYCH encefalopatiach na wolności. Badania genetyczne przeprowadzono u 85 osób, a 12/85 (14%) osób miało pozytywne wyniki genetyczne. U dwóch osobników znaleziono trzy geny, w tym SYNGAP1, NEXMIF (KIAA2022) i SLC6A1. Ta Triada genów była wcześniej powiązana z encefalopatią rozwojową i epileptyczną z napadami uogólnionymi. Ponadto stwierdzono pojedyncze osobniki z wariantami chorobotwórczymi w KCNA2, SCN2A, STX1B, KCNB1 i MECP2. Wszystkie te geny były wcześniej omawiane na naszym blogu i są znane z uogólnionych napadów i uogólnionych funkcji EEG.
brak genów w MAE. Podczas gdy niektóre wcześniejsze raporty znalazły warianty w SCN1A, nie znaleziono tego w naszej kohorcie. Ponadto, inne typowe geny padaczki są wyraźnie nieobecne, w tym CDKL5 i SLC2A1 (GLUT1). Sugeruje to, że krajobraz genetyczny MAE jest nieco odmienny i różni się od innych encefalopatii rozwojowych i EPILEPTYCZNYCH, mimo że tylko ograniczone wnioski można wyciągnąć ze stosunkowo małych kohort. Wydajność diagnostyczna w MAE nie jest bardzo wysoka-14% jest znacznie niższa niż zwykle obserwowana w encefalopatiach rozwojowych i EPILEPTYCZNYCH. Jednak wykluczając osoby o typowym rozwoju, plony diagnostyczne są znacznie wyższe, co sugeruje, że badania genetyczne mają wartość u dzieci z MAE. Ogólnie rzecz biorąc, nawet jeśli żaden osobnik z MAE nie miał wariantów chorobotwórczych w SLC2A1, badania genetyczne pod kątem uleczalnych przyczyn epilepsji genetycznych mogą być na ogół rozważane w atypowych uogólnionych epilepsjach.
zaginiona dziedziczność MAE. Jeśli badania genetyczne są negatywne dla MAE u ponad 80% osób, gdzie jest ukryte obciążenie genetyczne? Od 2020 r. proponuję dwa możliwe wyjaśnienia. Po pierwsze, obciążenie genetyczne uogólnionych epilepsji nie jest całkowicie nieznane. Ponad 30% populacji jest narażonych na takie schorzenia, jak padaczka z powodu nieobecności dzieci (CAE) lub młodzieńcza padaczka miokloniczna (JME). Jednak wyjaśnienie to nie jest podane przez przyczyny jednogenowe, ale przez ryzyko poligenowe, odnosząc się do addytywnego efektu tysięcy powszechnych wariantów genetycznych. Można wysnuć hipotezę, że MAE może reprezentować „skrajny fenotyp” uogólnionych epilepsji, gdzie to poligeniczne ryzyko jest szczególnie wysokie. Ta hipoteza jest sprawdzalna i na pewno będzie kontynuowana w przyszłości. W dodatku, jest więcej do ludzkiego genomu niż egzomu-ostatnie odkrycia w epilepsji podkreślają rolę niekodujących wariantów, takich jak powtórzenia ekspansji w rodzinnej dorosłej padaczce Mioklonicznej (FAME). Biorąc pod uwagę uderzająco podobne cechy kliniczne u wielu dzieci z MAE, to nadal może być uzasadnione szukać wspólnej genetycznej przyczyny dla wielu dzieci z Mae poza exome, przy użyciu sekwencjonowania całego genomu. Taking razem, oceniający poligenic risk scores (PRS) i cały genom sekwencjonowanie (WGS) są dwa potencjalni Aleje dalej utożsamiać genetyczną podstawę MAE.
co musisz wiedzieć. Zespół DOOSE ’ a (MAE) jest przypuszczalnie genetycznym zespołem padaczki o niejednorodnej przyczynie. Warianty chorobotwórcze występują u 14% dzieci i są zazwyczaj identyfikowane u dzieci z dodatkowymi cechami neurorozwojowymi, takimi jak opóźnienie rozwoju lub autyzm. Triada genów, w tym SYNGAP1, NEXMIF (KIAA2022) i SLC6A1, są nawracającymi przyczynami genetycznymi, które mogą wskazywać na wspólną podstawową biologię, która jest nieco odmienna od innych encefalopatii rozwojowych i EPILEPTYCZNYCH.

Ingo Helbig jest neurologiem dziecięcym i badaczem genetyki padaczki, pracującym w Children ’ s Hospital of Philadelphia (CHOP), USA. Prowadzi również grupę genetyki padaczki na Uniwersytecie w Kilonii w Niemczech.

