- połowa XIX wieku-1945: od roślin do pierwszych leków syntetycznychedytuj
- epinefryna, norepinefryna i amfetaminaedit
- Odkrycie i rozwój barbituracjedytuj
- InsulinEdit
- wczesne badania przeciwinfekcyjne: Salwarsan, Prontosil, penicylina i szczepieniaedytuj
- niebezpieczne leki i wczesne regulacje branżoweedytuj
- lata powojenne, 1945-1970edytuj
- dalsze postępy w badaniach przeciwinfekcyjnychedytuj
- rozwój i marketing leków przeciwnadciśnieniowychedytuj
- doustne środki Antykoncepcyjneedytuj
- zmiany w Talidomidzie i Kefauver-Harrisedytuj
- 1970–1980sEdit
- StatinsEdit
połowa XIX wieku-1945: od roślin do pierwszych leków syntetycznychedytuj
nowoczesny przemysł farmaceutyczny rozpoczął się od lokalnych aptekarzy, którzy rozszerzyli swoją tradycyjną rolę dystrybucji leków botanicznych, takich jak morfina i chinina, do produkcji hurtowej w połowie XIX wieku oraz od odkryć wynikających z badań stosowanych. Celowe odkrycie leków z roślin rozpoczęło się od izolacji w latach 1803-1805 morfiny – środka przeciwbólowego i pobudzającego sen-z opium przez niemieckiego asystenta aptekarza Friedricha Sertürnera, który nazwał ten związek na cześć greckiego boga snów, Morfeusza. Pod koniec lat osiemdziesiątych XIX wieku niemieccy producenci barwników udoskonalili oczyszczanie poszczególnych związków organicznych ze smoły i innych źródeł mineralnych, a także wprowadzili podstawowe metody w organicznej syntezie chemicznej. Rozwój syntetycznych metod chemicznych pozwolił naukowcom systematycznie zmieniać strukturę substancji chemicznych, a rozwój powstającej nauki farmakologicznej poszerzył ich zdolność do oceny biologicznych skutków tych zmian strukturalnych.
epinefryna, norepinefryna i amfetaminaedit
do 1890 roku odkryto głęboki wpływ ekstraktów nadnerczy na wiele różnych typów tkanek, rozpoczynając poszukiwania zarówno mechanizmu sygnalizacji chemicznej, jak i wysiłków na rzecz wykorzystania tych obserwacji do rozwoju nowych leków. Podniesienie ciśnienia krwi i zwężające naczynia działanie ekstraktów nadnerczy było szczególnie interesujące dla chirurgów jako środki hemostatyczne i leczenie wstrząsu, a wiele firm opracowało produkty oparte na ekstraktach nadnerczy zawierających różne czystości substancji czynnej. W 1897 roku John Abel z Johns Hopkins University zidentyfikował zasadę aktywną jako epinefrynę, którą wyizolował w stanie nieczystym jako sól siarczanową. Chemik przemysłowy jōkichi Takamine później opracował metodę otrzymywania epinefryny w stanie czystym i licencjonował tę technologię Parke-Davisowi. Parke-Davis sprzedawał epinefrynę pod nazwą handlową Adrenalin. Wstrzyknięta epinefryna okazała się szczególnie skuteczna w ostrym leczeniu ataków astmy, a wersja wziewna była sprzedawana w Stanach Zjednoczonych do 2011 roku (Primatene Mist). Do 1929 epinefryna została opracowana jako inhalator do stosowania w leczeniu przekrwienia błony śluzowej nosa.
chociaż jest wysoce skuteczny, Wymaganie wstrzyknięcia ograniczało stosowanie epinefryny i aktywnych po podaniu doustnym pochodnych. Strukturalnie podobny związek, efedryna (właściwie bardziej podobna do noradrenaliny), został zidentyfikowany przez japońskich chemików w roślinie Ma Huang i wprowadzony do obrotu przez Eli Lilly jako doustne leczenie astmy. Po pracy Henry 'ego Dale’ a i George ’ a Bargera w Burroughs-Wellcome, chemik akademicki Gordon Alles zsyntetyzował amfetaminę i przetestował ją u pacjentów z astmą w 1929 roku. Lek okazał się mieć tylko skromne działanie przeciw astmie, ale wywoływał odczucia radości i kołatania serca. Amfetamina została opracowana przez Smitha, Kline 'a i French’ a jako lek zmniejszający przekrwienie nosa pod nazwą handlową Benzedrine Inhaler. Amfetamina została ostatecznie opracowana w leczeniu narkolepsji, Post-encephalitycznego parkinsonizmu i podniesienia nastroju w depresji i innych wskazaniach psychiatrycznych. W 1937 r. został zatwierdzony przez American Medical Association jako nowy i nieoficjalny środek do tych zastosowań i pozostawał w powszechnym użyciu w depresji do czasu opracowania trójpierścieniowych leków przeciwdepresyjnych w latach 60.
Odkrycie i rozwój barbituracjedytuj

w 1903 roku Hermann Emil Fischer i Joseph von Mering ujawnili swoje odkrycie, że kwas dietylbarbiturowy, powstały w wyniku reakcji kwasu dietylomalonowego, tlenochlorku fosforu i mocznika, indukuje sen u psów. Odkrycie zostało opatentowane i licencjonowane przez Bayer pharmaceuticals, który sprzedawał związek pod nazwą handlową Veronal jako środek wspomagający sen od 1904 roku. Systematyczne badania wpływu zmian strukturalnych na potencję i czas działania doprowadziły do odkrycia fenobarbitalu w firmie Bayer w 1911 roku i odkrycia jego silnego działania przeciwpadaczkowego w 1912 roku. Fenobarbital był jednym z najczęściej stosowanych leków w leczeniu padaczki w latach 70., a od 2014 r. pozostaje na liście najważniejszych leków Światowej Organizacji Zdrowia. W latach 50. i 60. nastąpił wzrost świadomości na temat uzależniających właściwości i możliwości nadużywania barbituranów i amfetamin, co doprowadziło do zwiększenia ograniczeń w ich stosowaniu i rosnącego nadzoru rządowego nad lekarzami przepisującymi lek. Obecnie amfetamina jest w dużej mierze ograniczona do stosowania w leczeniu zaburzeń deficytu uwagi i fenobarbitalu w leczeniu padaczki.
InsulinEdit
seria eksperymentów przeprowadzonych od końca XIX wieku do początku XX wieku ujawniła, że cukrzyca jest spowodowana brakiem substancji normalnie wytwarzanej przez trzustkę. W 1869 Oskar Minkowski i Joseph von Mering odkryli, że cukrzyca może być wywołana u psów przez chirurgiczne usunięcie trzustki. W 1921 roku kanadyjski profesor Frederick Banting i jego student Charles Best powtórzyli to badanie i stwierdzili, że zastrzyki ekstraktu trzustkowego odwróciły objawy wytwarzane przez usunięcie trzustki. Wkrótce wykazano, że ekstrakt działa u ludzi, ale rozwój insulinoterapii jako rutynowej procedury medycznej został opóźniony przez trudności w produkcji materiału w wystarczającej ilości i powtarzalnej czystości. Naukowcy zwrócili się o pomoc do współpracowników przemysłowych w firmie Eli Lilly and Co. w oparciu o doświadczenie firmy w oczyszczaniu materiałów biologicznych na dużą skalę. Chemik George B. Walden z Eli Lilly and Company odkrył, że staranne dostosowanie pH ekstraktu pozwoliło na wytworzenie stosunkowo czystego gatunku insuliny. Pod naciskiem Uniwersytetu w Toronto i potencjalnym wyzwaniem Patentowym ze strony naukowców akademickich, którzy niezależnie opracowali podobną metodę oczyszczania, osiągnięto porozumienie w sprawie niewyłącznej produkcji insuliny przez wiele firm. Przed odkryciem i powszechną dostępnością insulinoterapii średnia długość życia diabetyków wynosiła tylko kilka miesięcy.
wczesne badania przeciwinfekcyjne: Salwarsan, Prontosil, penicylina i szczepieniaedytuj
rozwój leków do leczenia chorób zakaźnych był głównym celem wczesnych prac badawczo-rozwojowych; w 1900 r.zapalenie płuc, gruźlica i biegunka były trzema głównymi przyczynami zgonów w Stanach Zjednoczonych, a śmiertelność w pierwszym roku życia przekroczyła 10%.
w 1911 r.arfenamina, pierwszy syntetyczny lek przeciwinfekcyjny, została opracowana przez Paula Ehrlicha i chemika Alfreda Bertheima z Instytutu Terapii Doświadczalnej w Berlinie. Lek otrzymał nazwę handlową Salvarsan. Ehrlich, zwracając uwagę zarówno na ogólną toksyczność arsenu, jak i selektywną absorpcję niektórych barwników przez bakterie, wysunął hipotezę, że barwnik zawierający arsen o podobnych właściwościach selektywnej absorpcji może być stosowany w leczeniu zakażeń bakteryjnych. Arspenamina została przygotowana w ramach kampanii mającej na celu zsyntetyzowanie szeregu takich związków i stwierdzono, że wykazuje częściowo selektywną toksyczność. Arspenamina okazała się pierwszym skutecznym leczeniem kiły, choroby, która wcześniej była nieuleczalna i prowadziła nieubłaganie do ciężkich owrzodzeń skóry, uszkodzeń neurologicznych i śmierci.
podejście Ehrlicha do systematycznego zmieniania struktury chemicznej związków syntetycznych i mierzenia wpływu tych zmian na aktywność biologiczną było szeroko stosowane przez naukowców przemysłowych, w tym przez naukowców z firmy Bayer: Josefa Klarera, Fritza Mietzscha i Gerharda Domagka. Praca ta, również oparta na testowaniu związków dostępnych w niemieckim przemyśle barwników, doprowadziła do opracowania Prontosil, pierwszego przedstawiciela antybiotyków z grupy sulfonamidów. W porównaniu z arfenaminą sulfonamidy miały szersze spektrum aktywności i były znacznie mniej toksyczne, co czyni je przydatnymi w infekcjach wywoływanych przez patogeny, takie jak paciorkowce. W 1939 Domagk otrzymał za to odkrycie Nagrodę Nobla w dziedzinie medycyny. Niemniej jednak dramatyczny spadek liczby zgonów z powodu chorób zakaźnych, który miał miejsce przed II wojną światową, był przede wszystkim wynikiem poprawy środków w zakresie zdrowia publicznego, takich jak czysta woda i mniej zatłoczone mieszkania, a wpływ leków przeciwinfekcyjnych i szczepionek był znaczący głównie po ii Wojnie Światowej.
w 1928 roku Alexander Fleming odkrył przeciwbakteryjne działanie penicyliny, ale jej wykorzystanie do leczenia chorób ludzkich wymagało opracowania metod produkcji i oczyszczania na dużą skalę. Zostały one opracowane przez amerykańskie i brytyjskie rządowe konsorcjum firm farmaceutycznych podczas ii Wojny Światowej.
wczesny postęp w rozwoju szczepionek nastąpił w tym okresie, głównie w formie badań naukowych i finansowanych przez rząd badań podstawowych ukierunkowanych na identyfikację patogenów odpowiedzialnych za powszechne choroby zakaźne. W 1885 Louis Pasteur i Pierre Paul Émile Roux stworzyli pierwszą szczepionkę przeciwko wściekliźnie. Pierwsze szczepionki przeciw błonicy zostały wyprodukowane w 1914 roku z mieszaniny toksyny błonicy i antytoksyny (wytwarzanej z surowicy zaszczepionego zwierzęcia), ale bezpieczeństwo szczepienia było marginalne i nie było szeroko stosowane. W 1921 roku Stany Zjednoczone odnotowały 206 000 przypadków błonicy, w wyniku czego śmierć poniosło 15 520 osób. W 1923 roku równoległe wysiłki Gastona Ramona w Instytucie Pasteura i Alexandra Glenny ’ ego w Wellcome Research Laboratories (późniejsza część GlaxoSmithKline) doprowadziły do odkrycia, że bezpieczniejsza szczepionka może być wyprodukowana przez leczenie toksyny błonicy formaldehydem. W 1944 roku Maurice Hilleman z Squibb Pharmaceuticals opracował pierwszą szczepionkę przeciwko japońskiemu zapaleniu jelita grubego. Hilleman później przeniósł się do Merck, gdzie odegrał kluczową rolę w opracowywaniu szczepionek przeciwko odrze, śwince, ospie wietrznej, różyczce, wirusowemu zapaleniu wątroby typu A, wirusowemu zapaleniu wątroby typu B i zapaleniu opon mózgowych.
niebezpieczne leki i wczesne regulacje branżoweedytuj

przed 20 wieku, leki były na ogół produkowane przez producentów na małą skalę z niewielką kontrolą regulacyjną nad produkcją lub twierdzenia bezpieczeństwa i skuteczności. W zakresie, w jakim takie prawa istniały, egzekwowanie było luźne. W Stanach Zjednoczonych zwiększoną regulację szczepionek i innych leków biologicznych wywołały ogniska tężca i zgony spowodowane dystrybucją skażonej szczepionki przeciw ospie i antytoksyny błoniczej. Ustawa o kontroli biologicznej z 1902 r.wymagała, aby rząd federalny udzielił przedmarketowej zgody na każdy lek biologiczny oraz na proces i zakład produkujący takie leki. W 1906 roku ustawa Pure Food and Drugs Act zakazała międzypaństwowej dystrybucji zafałszowanej lub niezgodnej z marką żywności i narkotyków. Lek został uznany za niewłaściwy, jeśli zawierał alkohol, morfinę, opium, kokainę lub którykolwiek z kilku innych potencjalnie niebezpiecznych lub uzależniających leków, a jego etykieta nie wskazywała ilości lub proporcji takich leków. Próby rządu, aby wykorzystać prawo do ścigania producentów za składanie nieuzasadnionych roszczeń o skuteczność, zostały podcięte przez orzeczenie Sądu Najwyższego ograniczające uprawnienia wykonawcze rządu federalnego do przypadków nieprawidłowej specyfikacji składników leku.
w 1937 roku ponad 100 osób zmarło po spożyciu „eliksiru sulfanilamidu” produkowanego przez S. E. Massengill Company of Tennessee. Produkt został opracowany w glikolu dietylenowym, wysoce toksycznym rozpuszczalniku, który jest obecnie szeroko stosowany jako środek przeciw zamarzaniu. Zgodnie z obowiązującym wówczas prawem ściganie producenta było możliwe tylko pod warunkiem, że produkt został nazwany „eliksirem”, co dosłownie oznaczało rozwiązanie w etanolu. W odpowiedzi na ten epizod Kongres Stanów Zjednoczonych uchwalił federalną ustawę o żywności, lekach i kosmetykach z 1938 roku, która po raz pierwszy wymagała demonstracji bezpieczeństwa przed wprowadzeniem na rynek przed sprzedażą leku i wyraźnie zakazywała fałszywych twierdzeń terapeutycznych.
lata powojenne, 1945-1970edytuj
dalsze postępy w badaniach przeciwinfekcyjnychedytuj
po ii wojnie światowej nastąpił wybuch w odkryciu nowych klas leków przeciwbakteryjnych, w tym cefalosporyn (opracowanych przez Eli Lilly na podstawie przełomowych prac Giuseppe Brotzu i Edwarda Abrahama), streptomycyny (odkrytej podczas finansowanego przez Merck programu badawczego w Selman). laboratorium waksmana), tetracykliny (Odkryte w laboratoriach Lederle, obecnie część Pfizer), erytromycyna (odkryta w Eli Lilly and Co.) i ich rozszerzenie na coraz szerszy zakres patogenów bakteryjnych. Streptomycyna, odkryta podczas finansowanego przez Merck programu badawczego w laboratorium Selmana Waksmana w Rutgers w 1943 roku, stała się pierwszym skutecznym leczeniem gruźlicy. W momencie odkrycia, sanitariaty do izolacji osób zakażonych gruźlicą były wszechobecną cechą miast w krajach rozwiniętych, z 50% umiera w ciągu 5 lat od przyjęcia.
raport Federalnej Komisji Handlu wydany w 1958 roku próbował oszacować wpływ rozwoju antybiotyków na Amerykańskie zdrowie publiczne. W raporcie stwierdzono, że w okresie 1946-1955 nastąpił 42% spadek zachorowalności na choroby, w przypadku których antybiotyki były skuteczne, a tylko 20% spadek w przypadku chorób, w których antybiotyki nie były skuteczne. W raporcie stwierdzono, że”wydaje się, że stosowanie antybiotyków, wczesna diagnoza i inne czynniki ograniczyły rozprzestrzenianie się epidemii, a tym samym liczbę tych chorób, które wystąpiły”. W badaniu zbadano wskaźniki śmiertelności dla ośmiu powszechnych chorób, dla których antybiotyki oferowały skuteczną terapię (kiła, gruźlica, czerwonka, szkarlatyna, koklusz, zakażenia meningokokowe i zapalenie płuc) i stwierdzono 56% spadek w tym samym okresie. Zauważalny wśród nich był 75% spadek zgonów z powodu gruźlicy.
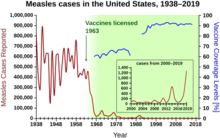
w latach 1940-1955 tempo spadku śmiertelności w USA przyspieszyło z 2% rocznie do 8% rocznie, a następnie powróciło do historycznej stopy 2% rocznie. Dramatyczny spadek w bezpośrednich latach powojennych został przypisany szybkiemu rozwojowi nowych metod leczenia i szczepionek na choroby zakaźne, które wystąpiły w tych latach.Rozwój szczepionek nadal przyspieszał, a najbardziej godnym uwagi osiągnięciem tego okresu było opracowanie przez Jonasa Salka szczepionki przeciw polio w 1954 roku, finansowanej przez Narodową Fundację non-profit na rzecz paraliżu infantylnego. Proces szczepionki nigdy nie został opatentowany, ale zamiast tego został przekazany firmom farmaceutycznym do produkcji jako tani lek generyczny. W 1960 Maurice Hilleman z Merck Sharp & zidentyfikował wirusa SV40, który później okazał się powodować nowotwory u wielu gatunków ssaków. Później ustalono, że SV40 był obecny jako zanieczyszczenie w partiach szczepionek przeciw polio, które zostały podane 90% dzieci w Stanach Zjednoczonych. Wydaje się, że zanieczyszczenie pochodzi zarówno z pierwotnego stada komórek, jak i z tkanek małp używanych do produkcji. W 2004 roku United States Cancer Institute ogłosił, że doszedł do wniosku, że SV40 nie jest związany z rakiem u ludzi.
inne godne uwagi nowe szczepionki tego okresu obejmują te na odrę (1962, John Franklin Enders z Children ’ s Medical Center Boston, później udoskonalone przez Maurice Hilleman w Merck), różyczkę (1969, Hilleman, Merck) i świnkę (1967, Hilleman, Merck) Stany Zjednoczone częstość występowania różyczki, wrodzonego zespołu różyczki, odry i świnki spadła o >95% w bezpośrednim następstwie powszechnego szczepienia. Pierwsze 20 lat licencjonowanego szczepienia przeciw odrze w USA zapobiegło około 52 milionom przypadków choroby, 17 400 przypadkach upośledzenia umysłowego i 5 200 zgonów.
rozwój i marketing leków przeciwnadciśnieniowychedytuj
nadciśnienie tętnicze jest czynnikiem ryzyka miażdżycy, niewydolności serca, choroby wieńcowej, udaru mózgu, choroby nerek i choroby tętnic obwodowych i jest najważniejszym czynnikiem ryzyka zachorowalności i śmiertelności z powodu chorób sercowo-naczyniowych w krajach uprzemysłowionych. Przed 1940 około 23% wszystkich zgonów wśród osób powyżej 50 roku życia było przypisanych nadciśnieniu tętniczemu. Ciężkie przypadki nadciśnienia były leczone chirurgicznie.
wczesne osiągnięcia w dziedzinie leczenia nadciśnienia obejmowały czwartorzędowe środki blokujące współczulny układ nerwowy jonów amonowych, ale związki te nigdy nie były szeroko stosowane ze względu na poważne skutki uboczne, ponieważ długoterminowe konsekwencje zdrowotne wysokiego ciśnienia krwi nie zostały jeszcze ustalone i ponieważ musiały być podawane przez wstrzyknięcie.
w 1952 roku naukowcy z Ciba odkryli pierwszy dostępny doustnie środek rozszerzający naczynia, hydralazynę. Główną wadą monoterapii hydralazyną było to, że traciła ona swoją skuteczność w czasie (tachyfilaksja). W połowie lat 50. Karl H. Beyer, James M. Sprague, John E. Baer i Frederick C. Novello z Merck and Co. odkrył i opracował chlorotiazyd, który pozostaje obecnie najczęściej stosowanym lekiem przeciwnadciśnieniowym. Rozwój ten był związany ze znacznym spadkiem śmiertelności wśród osób z nadciśnieniem tętniczym. Wynalazcy zostali wyróżnieni nagrodą Zdrowia Publicznego Laskera w 1975 roku za „uratowanie niezliczonych tysięcy istnień ludzkich i złagodzenie cierpienia milionów ofiar nadciśnienia”.
w przeglądzie Cochrane z 2009 r.stwierdzono, że tiazydowe leki przeciwnadciśnieniowe zmniejszają ryzyko zgonu (RR 0,89), udaru mózgu (RR 0,63), choroby wieńcowej serca (RR 0,84) i zdarzeń sercowo-naczyniowych (RR 0,70) u osób z wysokim ciśnieniem krwi. W kolejnych latach opracowano inne grupy leków przeciwnadciśnieniowych, które znalazły szerokie uznanie w terapii skojarzonej, w tym diuretyki pętlowe (Lasix/furosemid, Hoechst Pharmaceuticals, 1963), beta-adrenolityki (ICI Pharmaceuticals, 1964), inhibitory ACE i antagoniści receptora angiotensyny. Inhibitory ACE zmniejszają ryzyko wystąpienia nowej choroby nerek i zgonu u pacjentów z cukrzycą, niezależnie od tego, czy mają nadciśnienie.
doustne środki Antykoncepcyjneedytuj
przed II wojną światową kontrola urodzeń była zabroniona w wielu krajach,a w Stanach Zjednoczonych nawet dyskusja na temat metod antykoncepcyjnych czasami prowadziła do oskarżenia zgodnie z przepisami Comstock. Historia rozwoju doustnych środków antykoncepcyjnych jest zatem ściśle związana z ruchem kontroli urodzeń i wysiłkami aktywistów Margaret Sanger, Mary Dennett i Emmy Goldman. W oparciu o podstawowe badania przeprowadzone przez Gregory ’ ego Pincusa i syntetyczne metody progesteronu opracowane przez Carla Djerassi w Syntex i Franka Coltona w GD Searle & Co., pierwszy doustny środek antykoncepcyjny, Enovid, został opracowany przez E. D. Searle and Co. i zatwierdzony przez FDA w 1960 roku. Oryginalny preparat zawiera znacznie nadmierne dawki hormonów i spowodował poważne skutki uboczne. Niemniej jednak, do 1962 roku, 1,2 miliona amerykańskich kobiet było zażywających pigułki, a do 1965 roku liczba ta wzrosła do 6,5 miliona. Dostępność wygodnej formy tymczasowej antykoncepcji doprowadziła do dramatycznych zmian w obyczajach społecznych, w tym rozszerzenia zakresu opcji stylu życia dostępnych dla kobiet, zmniejszenia uzależnienia kobiet od mężczyzn w praktyce antykoncepcyjnej, zachęcania do opóźnienia małżeństwa i zwiększenia współżycia przedmałżeńskiego.
zmiany w Talidomidzie i Kefauver-Harrisedytuj

w USA, nacisk na rewizje ustawy FD& C wyłonił się z przesłuchań Kongresu prowadzonych przez senatora estesa Kefauvera z Tennessee w 1959 roku. Wysłuchania dotyczyły szerokiego zakresu kwestii politycznych, w tym nadużyć związanych z reklamą, wątpliwej skuteczności leków oraz potrzeby większej regulacji w branży. Podczas gdy impuls dla nowych przepisów był tymczasowo oznaczony w ramach rozszerzonej debaty, pojawiła się nowa tragedia, która podkreśliła potrzebę bardziej kompleksowej regulacji i stanowiła siłę napędową dla uchwalania nowych przepisów.
12 września 1960 urodził się Amerykański licencjat William S. Merrell Company of Cincinnati, złożyła wniosek o nowy lek na Kevadon (talidomid), środek uspokajający, który był wprowadzany do obrotu w Europie od 1956 roku. Oficer medyczny FDA odpowiedzialny za przegląd związku, Frances Kelsey, uważał, że dane potwierdzające bezpieczeństwo talidomidu są niekompletne. Firma nadal naciskała na Kelseya i FDA, aby zatwierdzili wniosek, aż do listopada 1961 roku, kiedy lek został wycofany z rynku niemieckiego z powodu jego związku z poważnymi wadami wrodzonymi. Kilka tysięcy noworodków w Europie i w innych krajach cierpiało na teratogenne działanie talidomidu. Bez zgody FDA firma rozprowadzała Kevadon do ponad 1000 lekarzy pod przykrywką eksperymentalnego zastosowania. Ponad 20 000 Amerykanów otrzymało talidomid w tym „badaniu”, w tym 624 kobiety w ciąży, a około 17 znanych noworodków cierpiało na działanie leku.
tragedia talidomidu wskrzesiła ustawę kefauvera o zaostrzeniu regulacji narkotyków, która utknęła w Kongresie, a poprawka Kefauvera-Harrisa stała się Ustawą 10 października 1962. Producenci musieli odtąd udowodnić FDA, że ich leki były skuteczne, a także bezpieczne, zanim mogli wejść na rynek amerykański. FDA otrzymała uprawnienia do regulowania reklamy leków na receptę i ustanowienia dobrych praktyk produkcyjnych. Prawo wymagało, aby wszystkie leki wprowadzone w latach 1938-1962 musiały być skuteczne. Wspólne badanie FDA-National Academy of Sciences wykazało, że prawie 40 procent tych produktów nie było skutecznych. Podobnie kompleksowe badanie produktów dostępnych bez recepty rozpoczęło się dziesięć lat później.
1970–1980sEdit
StatinsEdit
w 1971 roku Akira Endo, Japoński biochemik pracujący dla firmy farmaceutycznej Sankyo, zidentyfikował mewastatynę (ML-236B), cząsteczkę wytwarzaną przez grzyb Penicillium citrinum, jako inhibitor HMG-reduktaza Coa, krytyczny enzym stosowany przez organizm do produkcji cholesterolu. Badania na zwierzętach wykazały bardzo dobre działanie hamujące, podobnie jak w badaniach klinicznych, jednak długotrwałe badanie na psach wykazało działanie toksyczne po podaniu większych dawek, w wyniku czego mewastatyna była uważana za zbyt toksyczną dla ludzi. Mewastatyna nigdy nie został wprowadzony do obrotu, ze względu na jego niekorzystne skutki nowotworów, pogorszenie mięśni, a czasami śmierć u psów laboratoryjnych.
P. Roy Vagelos, główny naukowiec, a później dyrektor generalny Merck & Co, był zainteresowany i odbył kilka podróży do Japonii, począwszy od 1975 roku. W 1978 r. Merck wyizolował lowastatynę (mevinolin, MK803) z grzyba Aspergillus terreus, po raz pierwszy wprowadzoną do obrotu w 1987 r.jako Mevacor.
w kwietniu 1994 roku ogłoszono wyniki sponsorowanego przez Merck badania skandynawskiego badania przeżycia simwastatyny. Badacze przetestowali simwastatynę, sprzedawaną później przez Merck jako Zocor, na 4444 pacjentach z wysokim poziomem cholesterolu i chorobami serca. Po pięciu latach badania stwierdzono, że pacjenci odnotowali 35% zmniejszenie poziomu cholesterolu, a ich szanse na śmierć na atak serca zostały zmniejszone o 42%. W 1995 roku Zocor i Mevacor zarobiły ponad 1 miliard dolarów. Endo otrzymał w 2006 Japan Prize, a w 2008 Lasker-DeBakey Clinical Medical Research Award. Za „pionierskie badania nad nową klasą cząsteczek” za „obniżenie poziomu cholesterolu”