MAE. Existem muitas síndromes de epilepsia infantil distintas que nos tornamos criticamente conscientes na era genômica, pois estão ligadas a causas genéticas proeminentes, incluindo síndrome de Dravet (SCN1A) e epilepsia da Infância com crises focais migratórias (KCNT1). No entanto, existem muitas outras síndromes epilépticas onde uma causa genética tem sido suspeita há muito tempo, mas tem permanecido esquivo. Uma das síndromes epilépticas que permaneceu em grande parte inexplorada é a síndrome da Doose, também conhecida como epilepsia mioclónica Astática (MAE) ou epilepsia com crises mioclónicas-Atónicas. Em um estudo recente de Epilepsia, nós exploramos a arquitetura genética da Síndrome de Doose e identificados monogênica faz com que em 14% dos indivíduos, incluindo SYNGAP1, NEXMIF (KIAA2022), e SLC6A1. Nosso estudo sugere que a síndrome de Doose é geneticamente heterogênea, possivelmente com uma paisagem genética distinta.
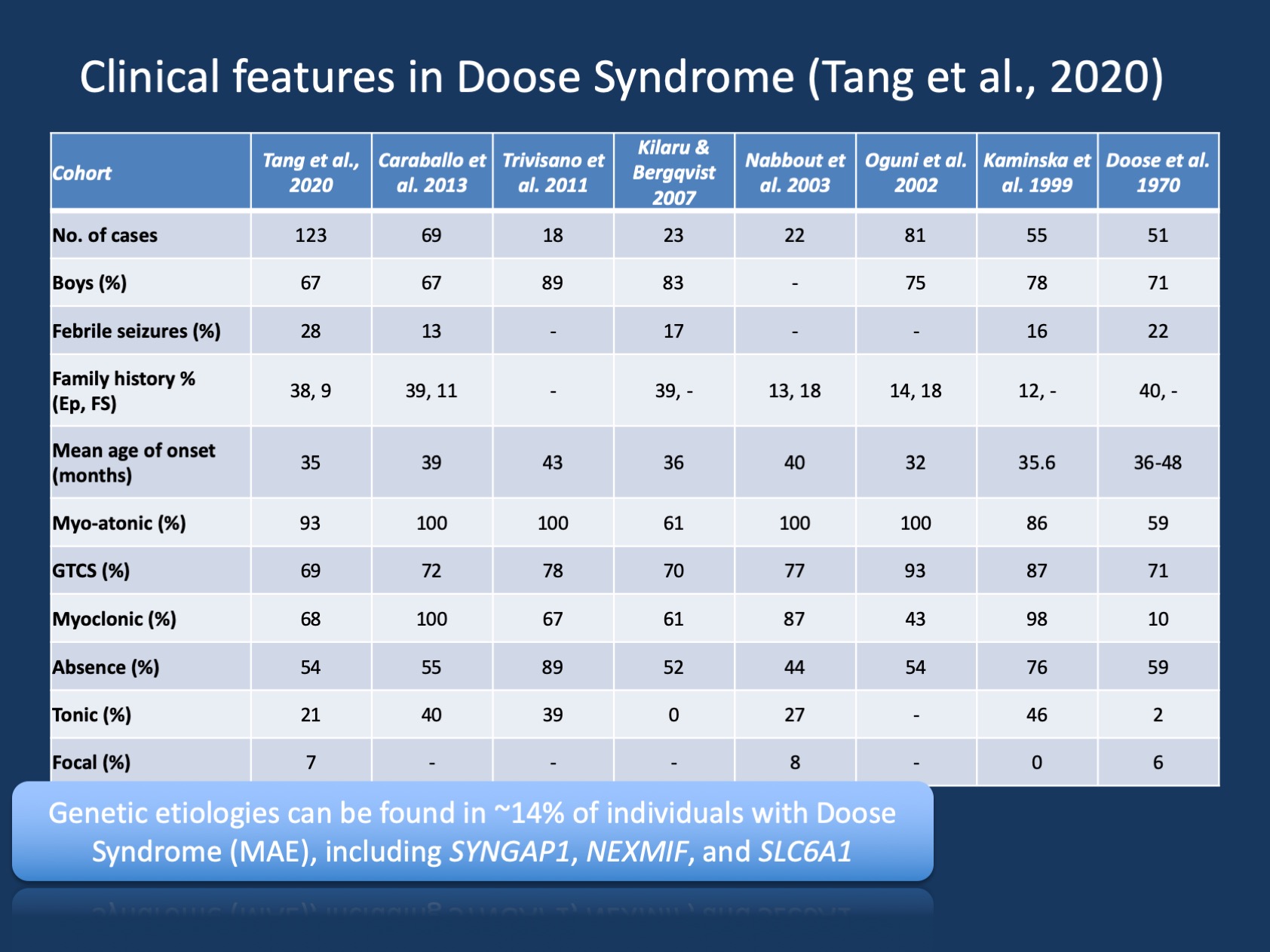
Figura 1. Características clínicas da síndrome de Doose (epilepsia com crises atónicas mioclónicas ou epilepsia Astática mioclónica ). A tabela é adaptada da nossa recente publicação de Tang et al., 2020 e compara as características clínicas das grandes coortes da síndrome da Doose que foram relatadas desde a descrição inicial em 1970. A síndrome de Doose (MAE) é uma síndrome epiléptica rara, mas é uma entidade clínica reconhecível. No entanto, em contraste com a síndrome de Dravet, onde >90% dos indivíduos têm variantes causadoras de doenças no SCN1A, a síndrome de Doose é geneticamente heterogênea.
a história de MAE. Em primeiro lugar, permitam-me que esclareça algumas questões relacionadas com nomes. Síndrome de Doose ou epilepsia Astática mioclónica (MAE) é agora oficialmente referida como epilepsia com crises atônicas mioclônicas usando a Convenção de nomenclatura mais recente. No entanto, enquanto normalmente tentamos alinhar-nos com a forma oficial de como os termos relacionados com a epilepsia são usados, deixe-me ser um pouco teimoso aqui. Como uma criança neurologista treinados no Departamento de Neuropediatria em Kiel, Alemanha, vou me referir a essa condição, como a Síndrome de Doose usando MAE como um atalho, honrando Hermann Doose (1927-2018), que é um dos fundadores do Europeu pediátrica epileptology (link para ILAE in memoriam). Muitos falantes nativos de Inglês tipicamente têm a pronúncia errada – a pronúncia correta é” Dohs-ah “e não rima com”ganso”.
o quadro clínico de MAE. O Doose é normalmente creditado pela realização de uma observação clínica importante.: na década de 1960, ele e seus colaboradores em Kiel reconheceram uma epilepsia incomum em crianças que foi caracterizada por ataques repentinos de queda (ataques atônicos), ataques mioclônicos, crises generalizadas tônico-clônicas, e um padrão generalizado de EEG, tipicamente ocorrendo entre os dois e cinco anos de idade. Esta epilepsia tipicamente ocorreu em crianças com desenvolvimento típico prévio (ver abaixo) e muitas vezes resultou em atraso no desenvolvimento e incapacidade intelectual. Doose e colaboradores reconheceram que esta síndrome de epilepsia recentemente reconhecida era diferente da síndrome de Lennox-Gastaut ou epilepsias com crises generalizadas tónico-clónicas precipitadas pela febre, a última das quais foi posteriormente conceitualizada como síndrome de Dravet.
Understanding MAE. As descrições clínicas da síndrome de Doose (MAE) têm sido constantes ao longo dos anos, mas MAE não recebeu tanta atenção como as síndromes de epilepsia infantil onde uma base genética tinha sido descoberta e onde pesquisas direcionadas podem permitir novos tratamentos. Em nossa recente publicação de Tang e colaboradores, temos listado oito estudos nos últimos 50 anos entre 1970 e 2020, com um total de 442 indivíduos descritos. Assim, MAE é rara, mas é comum o suficiente que a maioria dos neurologistas infantis e epileptólogos pediátricos estão cientes desta condição. Apenas um pequeno subconjunto de indivíduos com MAE tem achados cerebrais estruturais ou anomalias metabólicas que explicam a epilepsia subjacente – na grande maioria das crianças com MAE, neuroimaging e trabalho metabólico é normal.
encontrando a causa de MAE. A ausência de imagiologia explicativa ou de resultados metabólicos levanta a preocupação por uma causa genética subjacente. Embora estudos anteriores tenham tentado identificar uma contribuição genética através de estudos epidemiológicos, avaliando histórias familiares, este método tradicional tem sido largamente substituído por descobertas recentes em genética da epilepsia. A avaliação da base genética das epilepsias infantis graves é agora feita através da Genética molecular e não através da epidemiologia genética. Avaliar a arquitetura genética de MAE foi um dos objetivos em nosso estudo por Tang e colaboradores que foi publicado na Epilepsia.A MAE é uma doença? Antes de descrever os achados genéticos em MAE, deixe-me introduzir um debate em curso dentro do campo MAE. A descrição inicial por Doose e colaboradores incluiu em grande parte crianças com desenvolvimento típico antes do início das convulsões (>75%), O que é comparável ao nosso estudo recente (79%). No entanto, ao comparar as crianças com e sem problemas de desenvolvimento anteriores, estamos a olhar para a mesma doença ou só as crianças com desenvolvimento normal anterior devem ser consideradas MAE típicas? Embora esta questão não seja facilmente resolvida ao considerar as histórias e os resultados da epilepsia, as alterações genéticas subjacentes podem fornecer algumas pistas. As causas genéticas são identificadas com maior frequência em crianças com distúrbios de desenvolvimento neurológico mais graves e todas as crianças com causas genéticas identificadas tiveram distúrbios de desenvolvimento neurológico complexos, além da epilepsia. Assim, causas genéticas podem ser observadas em crianças com MAE, mas normalmente apenas em crianças com MAE e comorbidade com transtornos de neurodesenvolvimento, sugerindo que a MAE não é apenas um indivíduo geneticamente heterogêneas condição, mas tem características semelhantes como outros epilepsia na infância, com respeito à sua heterogeneidade genética e comorbidade. Por exemplo, na síndrome de West (espasmos infantis), as causas genéticas também são identificadas apenas em crianças com resultados de desenvolvimento neurológico e não em crianças que respondem diretamente à medicação sem sequelas de desenvolvimento neurológico.
descobertas genéticas em MAE. O espectro genético em MAE é diferente das encefalopatias epilépticas em geral. Os testes genéticos foram realizados em 85 indivíduos e 12/85 (14%) indivíduos tiveram resultados genéticos positivos. Três genes foram encontrados em dois indivíduos, incluindo SYNGAP1, NEXMIF (KIAA2022) e SLC6A1. Esta tríade de genes tem sido anteriormente ligada a encefalopatias epilépticas de desenvolvimento e com crises generalizadas. Além disso, indivíduos individuais foram encontrados com variantes causadoras de doenças em KCNA2, SCN2A, STX1B, KCNB1, e MECP2. Todos esses genes foram anteriormente discutidos em nosso blog e são conhecidos por apresentar convulsões generalizadas e recursos generalizados EEG.
genes ausentes em MAE. Embora alguns relatórios anteriores tenham encontrado variantes no SCN1A, isso não foi encontrado em nossa coorte. Além disso, outros genes epilépticos comuns estão notavelmente ausentes, incluindo CDKL5 e SLC2A1 (GLUT1). Isto sugere que a paisagem genética de MAE é um pouco distinta e diferente de outras encefalopatias epilépticas de desenvolvimento, embora apenas conclusões limitadas podem ser tiradas de coortes relativamente pequenas. O rendimento de diagnóstico em MAE não é muito alto-14% é muito menor do que normalmente visto nas encefalopatias epilépticas de desenvolvimento e epiléptico. No entanto, ao excluir indivíduos com desenvolvimento típico, os rendimentos diagnósticos é muito maior, sugerindo que o teste genético é de valor em crianças com MAE. Em geral, apesar de nenhum indivíduo com MAE ter variantes causadoras de doenças no SLC2A1, testes genéticos para causas tratáveis de epilepsias genéticas podem geralmente ser considerados em epilepsias generalizadas atípicas.
a falta de hereditariedade de MAE. Se o teste genético é negativo para MAE em mais de 80% dos indivíduos, onde está o fardo genético oculto? A partir de 2020, sugeriria duas explicações possíveis. Primeiro, a carga genética das epilepsias generalizadas não é totalmente desconhecida. É explicado mais de 30% do risco da população para situações como epilepsia da ausência Infantil (CAE) ou epilepsia mioclónica Juvenil (JME). No entanto, esta explicação não é dada por causas de gene único, mas por risco poligénico, referindo-se ao efeito aditivo de milhares de variantes genéticas comuns. It could be hypothesized that MAE may represent an “extreme phenotype” of generalized epilepsies where this polygenic risk is particularly high. Esta hipótese é testável e será definitivamente prosseguida no futuro. Além disso, há mais no genoma humano do que os achados exome – recentes nas epilepsias enfatizaram o papel das variantes não codificantes, como Expansões repetidas na epilepsia mioclônica adulta familiar (FAME). Dadas as características clínicas impressionantemente semelhantes em muitas crianças com MAE, ainda pode ser razoável procurar uma causa genética comum para muitas crianças com MAE fora do exome, usando sequenciamento do genoma inteiro. Considerando em conjunto, a avaliação das Pontuações de risco poligénico (PRS) e da sequenciação do genoma total (WGS) são duas vias potenciais para identificar ainda mais a base genética de MAE.O que precisa de saber. A síndrome de Doose (MAE) é uma síndrome epiléptica presumivelmente genética com uma causa heterogênea. Variantes causadoras de doenças estão presentes em 14% das crianças e são tipicamente identificadas em crianças com características de desenvolvimento neurológico adicionais, tais como atraso de desenvolvimento ou autismo. Uma tríade de genes, incluindo SYNGAP1, NEXMIF (KIAA2022), e SLC6A1, são causas genéticas recorrentes que podem sugerir uma biologia subjacente compartilhada que é um pouco diferente de outras encefalopatías de desenvolvimento e epiléptico.

Ingo Helbig é um neurologista infantil e pesquisador de genética da epilepsia que trabalha no Children’s Hospital de Filadélfia (CHOP), EUA. Ele também lidera o grupo de genética da epilepsia na Universidade de Kiel, Alemanha.

