MAE. Există multe sindroame distincte de Epilepsie din copilărie de care am devenit conștienți critic în epoca genomică, deoarece sunt legate de cauze genetice proeminente, inclusiv sindromul Dravet (SCN1A) și epilepsia copilăriei cu convulsii focale migratoare (KCNT1). Cu toate acestea, există multe alte sindroame de epilepsie în care o cauză genetică a fost suspectată de mult timp, dar a rămas evazivă. Unul dintre sindroamele epilepsiei care a rămas în mare parte neexplorat este sindromul Doose, denumit și epilepsie astatică mioclonică (MAE) sau epilepsie cu convulsii mioclonice-atonice. Într-un studiu recent privind Epilepsia, am explorat arhitectura genetică a sindromului Doose și am identificat cauze monogene la 14% dintre indivizi, inclusiv SYNGAP1, NEXMIF (KIAA2022) și SLC6A1. Studiul nostru sugerează că sindromul Doose este genetic eterogen, posibil cu un peisaj genetic distinct.
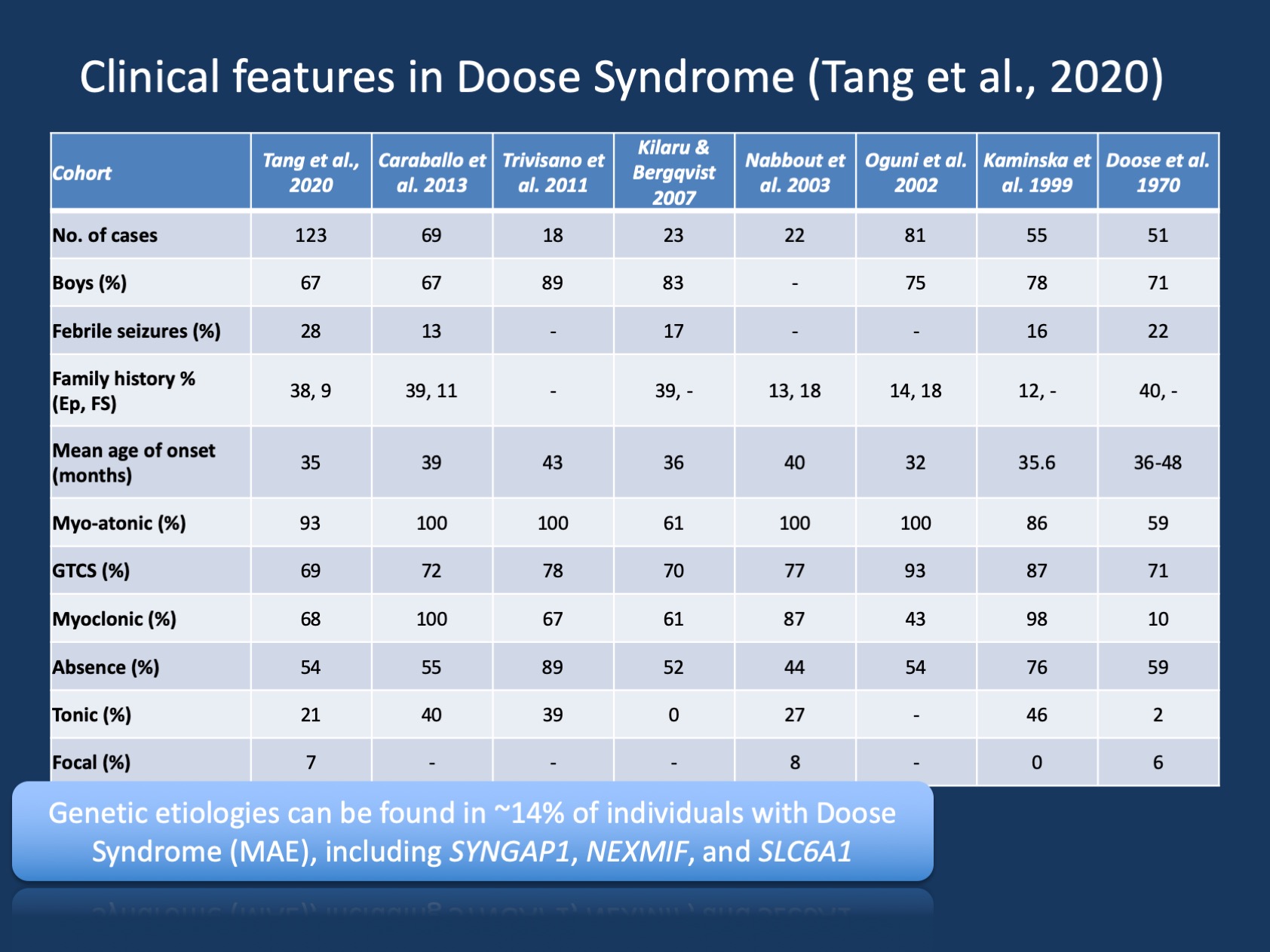
Figura 1. Caracteristicile clinice ale sindromului Doose (epilepsie cu convulsii atonice mioclonice sau epilepsie astatică mioclonică ). Tabelul este adaptat din publicația noastră recentă de Tang și colab., 2020 și compară caracteristicile clinice din cohortele mari de sindrom Doose care au fost raportate de la descrierea inițială din 1970. Sindromul Doose (MAE) este un sindrom de epilepsie rară, dar este o entitate clinică recunoscută. Cu toate acestea, spre deosebire de sindromul Dravet unde >90% dintre indivizi au variante cauzatoare de boli în SCN1A, sindromul Doose este eterogen genetic.
istoria MAE. În primul rând, permiteți-mi să clarific câteva probleme de denumire. Sindromul Doose sau epilepsia astatică mioclonică (MAE) este acum denumită oficial epilepsie cu convulsii mioclonice-atonice folosind cea mai recentă convenție de denumire. Cu toate acestea, în timp ce încercăm de obicei să ne aliniem cu modul oficial de utilizare a Termenilor legați de epilepsie, permiteți-mi să fiu puțin încăpățânat aici. În calitate de neurolog copil instruit la Departamentul de Neuropediatrie din Kiel, Germania, mă voi referi la această afecțiune ca sindrom Doose folosind MAE ca stenografie, onorându-l pe Hermann Doose (1927-2018) care este unul dintre fondatorii epileptologiei pediatrice Europene (link către ILAE in memoriam). Mulți vorbitori nativi de engleză greșesc de obicei pronunția – pronunția corectă este” Dohs-ah „și nu rimează cu”gâscă”.
tabloul clinic al MAE. Doose este de obicei creditat cu efectuarea unei observații clinice importante: în anii 1960, el și colaboratorii săi din Kiel au recunoscut o epilepsie neobișnuită la copii, care s-a caracterizat prin apariția bruscă a atacurilor de cădere (convulsii atonice), convulsii mioclonice, convulsii tonico-clonice generalizate și un model EEG generalizat, care apare de obicei între vârsta de doi și cinci ani. Această epilepsie a apărut de obicei la copiii cu o dezvoltare tipică anterioară (vezi mai jos) și a dus adesea la întârzierea dezvoltării și la dizabilități intelectuale. Doose și colaboratorii au recunoscut că acest sindrom de epilepsie nou recunoscut a fost diferit de sindromul Lennox-Gastaut sau epilepsiile cu convulsii tonico-clonice generalizate precipitate de febră, acestea din urmă fiind ulterior conceptualizate ca sindrom Dravet.
înțelegerea MAE. Descrierile clinice ale sindromului Doose (MAE) au fost constante de-a lungul anilor, dar MAE nu a primit la fel de multă atenție ca sindroamele de Epilepsie din copilărie în care a fost descoperită o bază genetică și unde cercetările vizate pot permite tratamente noi. În publicația noastră recentă realizată de Tang și colaboratori, am enumerat opt studii în ultimii 50 de ani între 1970 și 2020, cu un total de 442 de persoane descrise. În consecință, MAE este rar, dar este destul de comun ca majoritatea neurologilor copiilor și epileptologilor pediatrici să fie conștienți de această afecțiune. Doar un mic subset de indivizi cu MAE au constatări structurale ale creierului sau anomalii metabolice care explică epilepsia subiacentă – în marea majoritate a copiilor cu MAE, neuroimagistica și activitatea metabolică sunt de neimaginat.
găsirea cauzei MAE. Absența imagisticii explicative sau a constatărilor metabolice ridică îngrijorarea pentru o cauză genetică subiacentă. În timp ce studiile anterioare au încercat să se bazeze pe o contribuție genetică prin studii epidemiologice prin evaluarea Istoriilor familiale, această metodă tradițională a fost în mare parte înlocuită de descoperirile recente în genetica epilepsiei. Evaluarea bazei genetice a epilepsiilor severe din copilărie se face acum prin genetică moleculară, mai degrabă decât prin Epidemiologie genetică. Evaluarea arhitecturii genetice a MAE a fost unul dintre obiectivele studiului nostru realizat de Tang și colaboratori care a fost publicat în Epilepsia.
este MAE o boală? Înainte de a descrie constatările genetice în MAE, permiteți-mi să introduc o dezbatere în curs în domeniul MAE. Descrierea inițială de către Doose și colaboratori a inclus în mare parte copii cu dezvoltare tipică înainte de debutul convulsiilor (>75%), ceea ce este comparabil cu studiul nostru recent (79%). Cu toate acestea, atunci când comparăm copiii cu și fără probleme de dezvoltare anterioare, ne uităm la aceeași boală sau ar trebui considerați doar copiii cu o dezvoltare anterioară neremarcabilă MAE tipic? Deși această întrebare nu este ușor de rezolvat atunci când se iau în considerare istoriile și rezultatele epilepsiei, modificările genetice care stau la baza pot oferi unele indicii. Cauzele genetice sunt identificate mai frecvent la copiii cu tulburări neurodevelopmentale mai severe și toți copiii cu cauze genetice identificate au avut tulburări neurodevelopmentale complexe în plus față de epilepsie. În consecință, cauzele genetice pot fi observate la copiii cu MAE, dar de obicei numai la copiii cu MAE și tulburări de neurodezvoltare comorbide, sugerând că MAE nu este doar o afecțiune eterogenă genetic, ci are caracteristici similare cu alte epilepsii din copilărie în ceea ce privește eterogenitatea și comorbiditatea lor genetică. De exemplu, în sindromul West (spasme Infantile), cauzele genetice sunt, de asemenea, identificate numai la copiii cu constatări neurodezvoltate și nu la copiii care răspund direct la medicamente fără sechele neurodezvoltate.
descoperiri genetice în MAE. Spectrul genetic în MAE este diferit de cel al encefalopatiilor epileptice în general. Testarea genetică a fost efectuată la 85 de persoane, iar 12/85 (14%) au avut rezultate genetice pozitive. Trei gene au fost găsite la doi indivizi, inclusiv SYNGAP1, NEXMIF (KIAA2022) și SLC6A1. Această triadă de gene a fost legată anterior de encefalopatii de dezvoltare și epileptice cu convulsii generalizate. În plus, au fost găsiți indivizi singuri cu variante cauzatoare de boli în KCNA2, SCN2A, STX1B, KCNB1 și MECP2. Toate aceste gene au fost discutate anterior pe blogul nostru și se știe că prezintă convulsii generalizate și caracteristici EEG generalizate.
gene absente în MAE. În timp ce unele rapoarte anterioare au găsit variante în SCN1A, acest lucru nu a fost găsit în cohorta noastră. În plus, alte gene comune de epilepsie sunt absente, inclusiv CDKL5 și SLC2A1 (GLUT1). Acest lucru sugerează că peisajul genetic al MAE este oarecum distinct și diferit de alte encefalopatii de dezvoltare și epileptice, chiar dacă se pot trage doar concluzii limitate din cohorte relativ mici. Randamentul diagnostic în MAE nu este foarte mare – 14% este mult mai mic decât se observă în mod obișnuit în encefalopatiile de dezvoltare și epileptice. Cu toate acestea, atunci când excludem persoanele cu dezvoltare tipică, randamentele diagnostice sunt mult mai mari, sugerând că testarea genetică este de valoare la copiii cu MAE. În general, chiar dacă nici un individ cu MAE nu a avut variante cauzatoare de boală în SLC2A1, testarea genetică pentru cauzele tratabile ale epilepsiilor genetice poate fi luată în considerare în general în epilepsiile generalizate atipice.
lipsa heritabilității MAE. Dacă testarea genetică este negativă pentru MAE la mai mult de 80% dintre indivizi, unde este povara genetică ascunsă? Începând cu 2020, aș sugera două explicații posibile. În primul rând, povara genetică a epilepsiilor generalizate nu este complet necunoscută. Se explică mai mult de 30% din riscul populației pentru afecțiuni precum epilepsia absenței copilăriei (CAE) sau epilepsia mioclonică juvenilă (JME). Cu toate acestea, această explicație nu este dată de cauzele cu o singură genă, ci de riscul poligenic, referindu-se la efectul aditiv al mii de variante genetice comune. S-ar putea presupune că MAE poate reprezenta un „fenotip extrem” al epilepsiilor generalizate în care acest risc poligenic este deosebit de ridicat. Această ipoteză este testabilă și va fi cu siguranță urmărită în viitor. În plus, există mai mult pentru genomul uman decât exomul – descoperirile recente din epilepsii au subliniat rolul variantelor necodificatoare, cum ar fi extinderile repetate în epilepsia mioclonică familială adultă (FAME). Având în vedere caracteristicile clinice izbitor de similare la mulți copii cu MAE, poate fi totuși rezonabil să căutăm o cauză genetică comună pentru mulți copii cu MAE în afara exomului, folosind secvențierea întregului genom. Luând împreună, evaluarea scorurilor de risc poligenic (PRS) și secvențierea întregului genom (WGS) sunt două căi potențiale pentru a identifica în continuare baza genetică a MAE.
ce trebuie să știți. Sindromul Doose (MAE) este un sindrom de epilepsie probabil genetic cu o cauză eterogenă. Variantele cauzatoare de boli sunt prezente la 14% dintre copii și sunt de obicei identificate la copiii cu caracteristici suplimentare de neurodezvoltare, cum ar fi întârzierea dezvoltării sau autismul. O triadă de gene, inclusiv SYNGAP1, NEXMIF (KIAA2022) și SLC6A1, sunt cauze genetice recurente care pot sugera o biologie comună care este oarecum distinctă de alte encefalopatii de dezvoltare și epileptice.

Ingo Helbig este neurolog pentru copii și cercetător în genetica epilepsiei care lucrează la Spitalul de copii din Philadelphia (CHOP), SUA. De asemenea, conduce grupul de genetică pentru epilepsie de la Universitatea din Kiel, Germania.

