MAE. Der er mange forskellige barndomsepilepissyndromer, som vi er blevet kritisk opmærksomme på i den genomiske æra, da de er knyttet til fremtrædende genetiske årsager, herunder Dravet syndrom (SCN1A) og epilepsi i barndommen med migrerende fokale anfald (KCNT1). Der er dog mange andre epilepsisyndromer, hvor en genetisk årsag længe har været mistænkt, men har været undvigende. Et af de epilepsisyndromer, der stort set har været uudforsket, er Doose-syndrom, også kaldet myoklonisk astatisk epilepsi (MAE) eller epilepsi med myoklonisk-atoniske anfald. I en nylig undersøgelse i epilepsi udforskede vi den genetiske arkitektur af Doose syndrom og identificerede monogene årsager hos 14% af individer, herunder SYNGAP1, næste hvis (KIAA2022) og SLC6A1. Vores undersøgelse antyder, at Doose syndrom er genetisk heterogent, muligvis med et særskilt genetisk landskab.
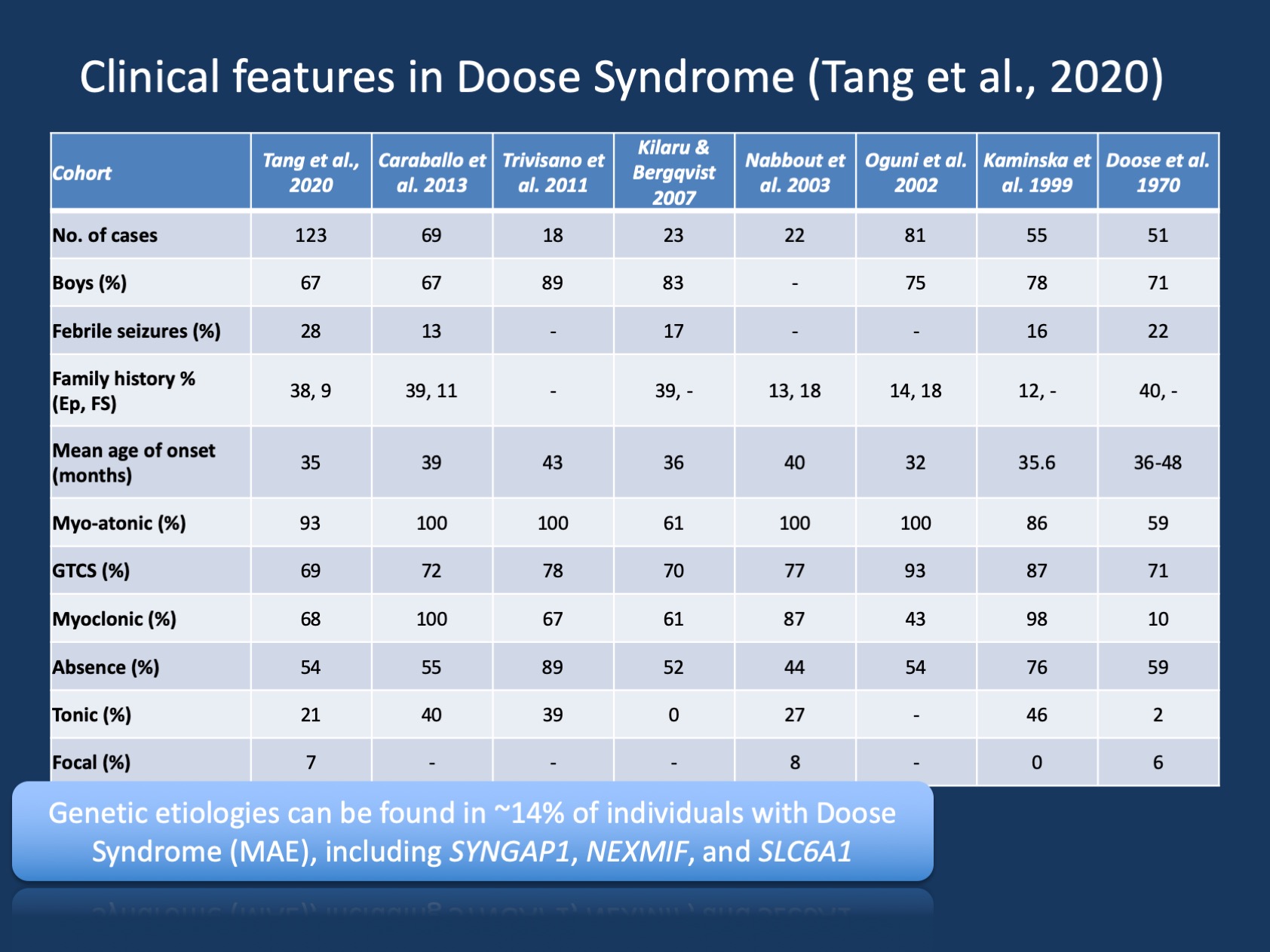
Figur 1. De kliniske træk ved Doose syndrom (epilepsi med myokloniske atoniske anfald eller myoklonisk astatisk epilepsi ). Tabellen er tilpasset fra vores nylige publikation af Tang et al., 2020 og sammenligner de kliniske træk i de store kohorter af Doose syndrom, der er rapporteret siden den første beskrivelse i 1970. Doose syndrom (MAE) er et sjældent epilepsisyndrom, men er en genkendelig klinisk enhed. I modsætning til Dravet-syndrom, hvor >90% af individer har sygdomsfremkaldende varianter i SCN1A, er Doose-syndrom genetisk heterogent.
Mae ‘ s historie. Først og fremmest, lad mig rydde op nogle navngivningsproblemer. Doose syndrom eller myoklonisk astatisk epilepsi (MAE) kaldes nu officielt epilepsi med myoklonisk-atoniske anfald ved hjælp af den seneste navngivningskonvention. Imidlertid, mens vi typisk forsøger at tilpasse os den officielle måde, hvorpå epilepsirelaterede udtryk bruges, lad mig være lidt stædig her. Som børne neurolog uddannet ved Institut for Neuropediatri i Kiel, Tyskland, vil jeg henvise til denne tilstand som Doose syndrom ved hjælp af MAE som en stenografi, ære Hermann Doose (1927-2018), som er en af grundlæggerne af europæisk pædiatrisk epileptologi (link til ILAE in memoriam). Mange indfødte engelsktalende får typisk udtalen forkert – den korrekte udtale er “Dohs-ah” og rimer ikke med “gås”.
det kliniske billede af MAE. Doose krediteres typisk med at foretage en vigtig klinisk observation: i 1960 ‘ erne anerkendte han og hans samarbejdspartnere i Kiel en usædvanlig epilepsi hos børn, der var kendetegnet ved pludselig begyndelse af faldangreb (atoniske anfald), myokloniske anfald, generaliserede tonisk-kloniske anfald og et generaliseret EEG-mønster, der typisk forekommer mellem to og fem år. Denne epilepsi forekom typisk hos børn med tidligere typisk udvikling (se nedenfor) og resulterede ofte i udviklingsforsinkelse og intellektuel handicap. Doose og samarbejdspartnere erkendte, at dette nyligt anerkendte epilepsisyndrom var forskelligt fra Lennoks-Gastaut syndrom eller epilepsier med generaliserede tonisk-kloniske anfald udfældet af feber, hvoraf sidstnævnte efterfølgende blev konceptualiseret som Dravet syndrom.
forståelse MAE. Kliniske beskrivelser af Doose syndrom (MAE) har været stabile gennem årene, men MAE har ikke fået så meget opmærksomhed som barndomsepilepissyndromer, hvor et genetisk grundlag var blevet opdaget, og hvor målrettet forskning kan muliggøre nye behandlinger. I vores nylige publikation af Tang og samarbejdspartnere har vi listet otte undersøgelser i de sidste 50 år mellem 1970 og 2020 med i alt 442 beskrevne personer. Derfor er MAE sjælden, men det er almindeligt nok, at de fleste børneurologer og pædiatriske epileptologer er opmærksomme på denne tilstand. Kun en lille delmængde af personer med MAE har strukturelle hjernefund eller metaboliske abnormiteter, der forklarer den underliggende epilepsi – hos langt de fleste børn med MAE er neuroimaging og metabolisk oparbejdning ikke bemærkelsesværdig.
Find årsagen til MAE. Fraværet af forklarende billeddannelse eller metaboliske fund rejser bekymring for en underliggende genetisk årsag. Mens tidligere undersøgelser havde forsøgt at fastlægge et genetisk bidrag gennem epidemiologiske undersøgelser ved at vurdere familiehistorier, denne traditionelle metode er stort set blevet afløst af nylige opdagelser inden for epilepsigenetik. Vurdering af det genetiske grundlag for alvorlige epilepsier i barndommen sker nu gennem molekylær genetik snarere end genetisk epidemiologi. Vurdering af Mae ‘ s genetiske arkitektur var et af målene i Vores undersøgelse af Tang og samarbejdspartnere, der blev offentliggjort i Epilepsia.
er MAE en sygdom? Før jeg beskriver de genetiske fund i MAE, lad mig introducere en løbende debat inden for MAE-feltet. Den indledende beskrivelse af Doose og samarbejdspartnere omfattede stort set børn med typisk udvikling inden anfaldsdebut (>75%), hvilket kan sammenlignes med vores nylige undersøgelse (79%). Men når vi sammenligner børn med og uden forudgående udviklingsmæssige problemer, ser vi på den samme sygdom, eller bør kun børn med forudgående unremarkable udvikling betragtes som typisk MAE? Selvom dette spørgsmål ikke let løses, når man overvejer epilepsihistorierne og resultaterne, kan de underliggende genetiske ændringer give nogle spor. Genetiske årsager identificeres hyppigere hos børn med mere alvorlige neuroudviklingsforstyrrelser, og alle børn med identificerede genetiske årsager havde komplekse neuroudviklingsforstyrrelser ud over epilepsi. Følgelig kan genetiske årsager observeres hos børn med MAE, men typisk kun hos børn med MAE og comorbide neuroudviklingsforstyrrelser, hvilket antyder, at MAE ikke kun er en genetisk heterogen tilstand, men har lignende træk som anden epilepsi i barndommen med hensyn til deres genetiske heterogenitet og comorbiditet. For eksempel i Vestsyndrom (Infantile spasmer) identificeres genetiske årsager også kun hos børn med neuroudviklingsresultater og ikke hos børn, der direkte reagerer på medicin uden neuroudviklingsfølger.
genetiske fund i MAE. Det genetiske spektrum i MAE er anderledes end i de epileptiske encephalopatier som helhed. Genetisk test blev udført hos 85 individer, og 12/85 (14%) individer havde positive genetiske fund. Tre gener blev fundet hos to individer, herunder SYNGAP1, NIF (KIAA2022) og SLC6A1. Denne triade af gener har tidligere været forbundet med udviklingsmæssige og epileptiske encephalopatier med generaliserede anfald. Derudover blev enkelte individer fundet med sygdomsfremkaldende varianter i KCNA2, SCN2A, STKS1B, KCNB1 og MECP2. Alle disse gener blev tidligere diskuteret på vores blog og er kendt for at præsentere med generaliserede anfald og generaliserede EEG-funktioner.
fraværende gener i MAE. Mens nogle tidligere rapporter har fundet varianter i SCN1A, blev dette ikke fundet i vores kohorte. Derudover er andre almindelige epilepsigener især fraværende, herunder CDKL5 og SLC2A1 (GLUT1). Dette antyder, at det genetiske landskab af MAE er noget forskelligt og forskelligt fra andre udviklings-og epileptiske encephalopatier, selvom kun begrænsede konklusioner kan drages fra relativt små kohorter. Det diagnostiske udbytte i MAE er ikke særlig højt – 14% er meget lavere end typisk set i udviklings-og epileptiske encephalopatier. Men når man udelukker personer med typisk udvikling, er de diagnostiske udbytter meget højere, hvilket tyder på, at genetisk testning er af værdi hos børn med MAE. Generelt, selvom ingen enkeltperson med MAE havde sygdomsfremkaldende varianter i SLC2A1, genetisk test for behandlingsårsager til genetiske epilepsier kan generelt overvejes i atypiske generaliserede epilepsier.
Mae ‘ s manglende arvelighed. Hvis genetisk test er negativ for MAE hos mere end 80% af individerne, hvor er den skjulte genetiske byrde? Fra 2020 vil jeg foreslå to mulige forklaringer. For det første er den genetiske byrde af generaliserede epilepsier ikke helt ukendt. Mere end 30% af befolkningens risiko for tilstande som f.eks barndom fravær epilepsi (CAE) eller juvenil myoklonisk epilepsi (JME) forklares. Denne forklaring gives imidlertid ikke af enkeltgenårsager, men af polygenisk risiko, der henviser til den additive virkning af tusinder af almindelige genetiske varianter. Det kunne antages, at MAE kan repræsentere en “ekstrem fænotype” af generaliserede epilepsier, hvor denne polygene risiko er særlig høj. Denne hypotese er testbar og vil helt sikkert blive forfulgt i fremtiden. Derudover er der mere ved det menneskelige genom end eksomet – nylige fund i epilepsierne har understreget rollen som ikke-kodende varianter, såsom gentagne udvidelser i familiær voksen myoklonisk epilepsi (FAME). I betragtning af de slående lignende kliniske træk hos mange børn med MAE, det kan stadig være rimeligt at lede efter en fælles genetisk årsag for mange børn med MAE uden for eksomet, ved hjælp af helgenomsekventering. Samlet set er vurdering af polygeniske risikoscorer (PRS) og helgenomsekventering to potentielle veje til yderligere at identificere det genetiske grundlag for MAE.
hvad du behøver at vide. Doose syndrom (MAE) er et formodentlig genetisk epilepsisyndrom med en heterogen årsag. Sygdomsfremkaldende varianter er til stede hos 14% af børnene og identificeres typisk hos børn med yderligere neuroudviklingsfunktioner såsom udviklingsforsinkelse eller autisme. En triade af gener, herunder SYNGAP1, NIF (KIAA2022) og SLC6A1, er tilbagevendende genetiske årsager, der kan antyde en fælles underliggende biologi, der er noget forskellig fra andre udviklings-og epileptiske encephalopatier.

Ingo Helbig er en børne neurolog og epilepsi genetik forsker, der arbejder på Children ‘ s Hospital of Philadelphia (CHOP), USA. Han leder også gruppen epilepsi genetik ved Universitetet i Kiel, Tyskland.

