MAE. Es gibt viele verschiedene Epilepsiesyndrome im Kindesalter, die uns in der genomischen Ära kritisch bewusst geworden sind, da sie mit prominenten genetischen Ursachen verbunden sind, einschließlich Dravet-Syndrom (SCN1A) und Epilepsie der Kindheit mit migrierenden fokalen Anfällen (KCNT1). Es gibt jedoch viele andere Epilepsiesyndrome, bei denen eine genetische Ursache lange vermutet wurde, aber schwer fassbar blieb. Eines der weitgehend unerforschten Epilepsiesyndrome ist das Doose-Syndrom, auch als myoklonische astatische Epilepsie (MAE) oder Epilepsie mit myoklonisch-atonischen Anfällen bezeichnet. In einer kürzlich durchgeführten Studie zu Epilepsie untersuchten wir die genetische Architektur des Doose-Syndroms und identifizierten bei 14% der Personen monogene Ursachen, darunter SYNGAP1, NEXMIF (KIAA2022) und SLC6A1. Unsere Studie legt nahe, dass das Doose-Syndrom genetisch heterogen ist, möglicherweise mit einer ausgeprägten genetischen Landschaft.
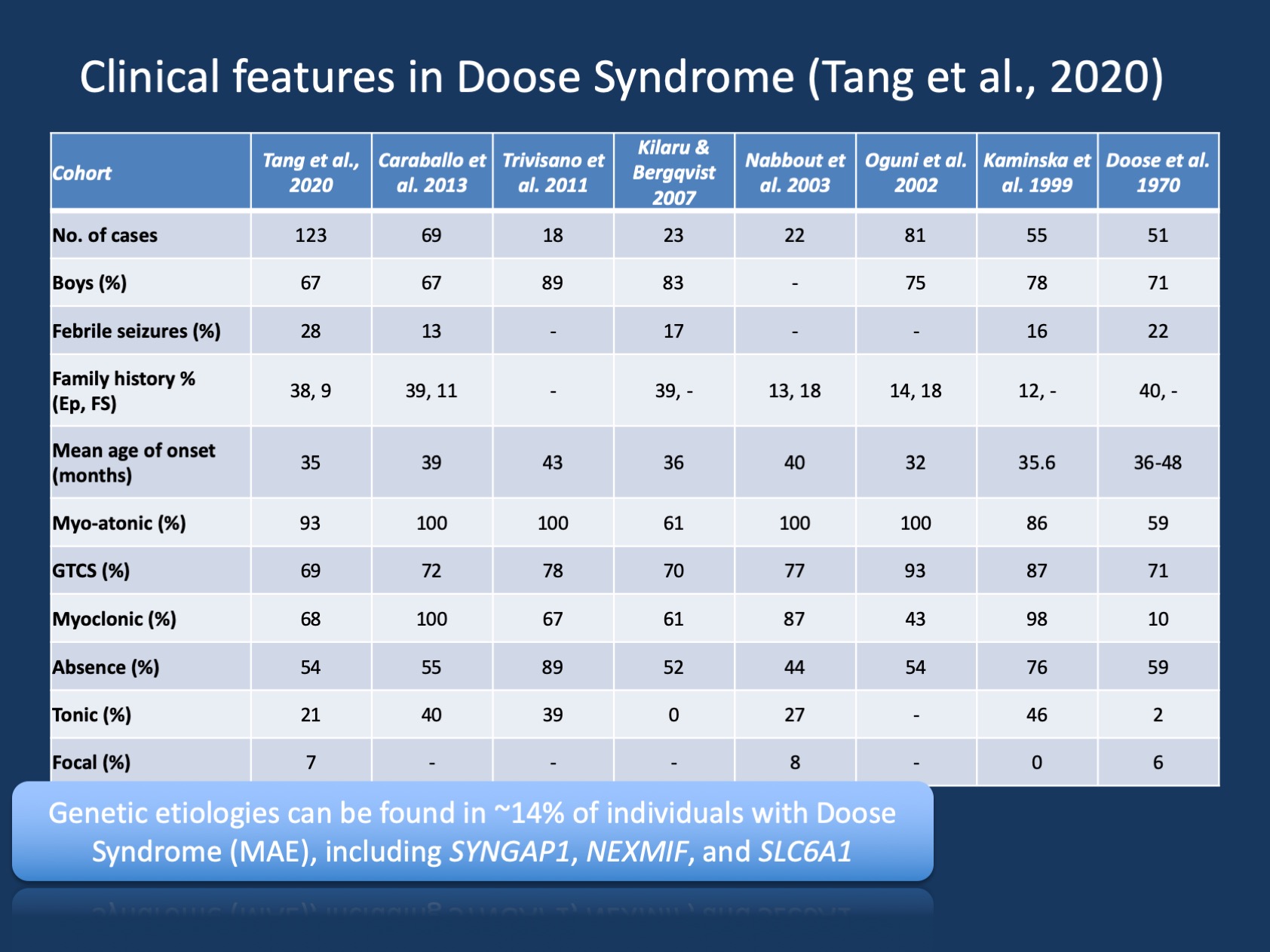
Abbildung 1. Die klinischen Merkmale des Doose-Syndroms (Epilepsie mit myoklonischen atonischen Anfällen oder myoklonischer astatischer Epilepsie ). Die Tabelle ist aus unserer jüngsten Veröffentlichung von Tang et al., 2020 und vergleicht die klinischen Merkmale in den großen Kohorten des Doose-Syndroms, die seit der Erstbeschreibung im Jahr 1970 berichtet wurden. Das Doose-Syndrom (MAE) ist ein seltenes Epilepsiesyndrom, aber eine erkennbare klinische Einheit. Im Gegensatz zum Dravet-Syndrom, bei dem > 90% der Individuen krankheitsverursachende Varianten in SCN1A aufweisen, ist das Doose-Syndrom jedoch genetisch heterogen.
Die Geschichte von MAE. Lassen Sie mich zunächst einige Namensfragen klären. Doose-Syndrom oder myoklonische astatische Epilepsie (MAE) wird jetzt offiziell als Epilepsie mit myoklonisch-atonischen Anfällen unter Verwendung der neuesten Namenskonvention bezeichnet. Während wir jedoch normalerweise versuchen, uns an die offizielle Art und Weise anzupassen, wie Epilepsie-bezogene Begriffe verwendet werden, lassen Sie mich hier etwas stur sein. Als an der Klinik für Neuropädiatrie in Kiel ausgebildeter Kinderneurologe werde ich diesen Zustand mit MAE als Abkürzung als Doose-Syndrom bezeichnen und Hermann Doose (1927-2018) ehren, der einer der Begründer der europäischen pädiatrischen Epileptologie ist (Link zu ILAE in memoriam). Viele englische Muttersprachler verstehen die Aussprache normalerweise falsch – die korrekte Aussprache ist „Dohs-ah“ und reimt sich NICHT auf „Gans“.
Das klinische Bild von MAE. Doose wird typischerweise eine wichtige klinische Beobachtung zugeschrieben: in den 1960er Jahren erkannten er und seine Mitarbeiter in Kiel eine ungewöhnliche Epilepsie bei Kindern, die durch plötzlich einsetzende Tropfenattacken (atonische Anfälle), myoklonische Anfälle, generalisierte tonisch-klonische Anfälle und ein generalisiertes EEG-Muster gekennzeichnet war, das typischerweise zwischen dem Alter von zwei und fünf Jahren auftrat. Diese Epilepsie trat typischerweise bei Kindern mit vorheriger typischer Entwicklung auf (siehe unten) und führte häufig zu Entwicklungsverzögerungen und geistiger Behinderung. Doose und Mitarbeiter erkannten, dass sich dieses neu erkannte Epilepsiesyndrom vom Lennox-Gastaut-Syndrom oder Epilepsien mit generalisierten tonisch-klonischen Anfällen, die durch Fieber ausgelöst wurden, unterschied, von denen letztere später als Dravet-Syndrom konzipiert wurden.
MAE verstehen. Klinische Beschreibungen des Doose-Syndroms (MAE) waren im Laufe der Jahre stabil, aber MAE hat nicht so viel Aufmerksamkeit erhalten wie Epilepsiesyndrome im Kindesalter, bei denen eine genetische Grundlage entdeckt wurde und bei denen gezielte Forschung möglicherweise neuartige Behandlungen ermöglicht. In unserer aktuellen Publikation von Tang und Mitarbeitern haben wir acht Studien der letzten 50 Jahre zwischen 1970 und 2020 mit insgesamt 442 beschriebenen Personen aufgelistet. Dementsprechend ist MAE selten, aber es ist üblich, dass die meisten Kinderneurologen und pädiatrischen Epileptologen sich dieser Erkrankung bewusst sind. Nur eine kleine Untergruppe von Personen mit MAE hat strukturelle Gehirnbefunde oder Stoffwechselstörungen, die die zugrunde liegende Epilepsie erklären – bei der überwiegenden Mehrheit der Kinder mit MAE sind Neuroimaging und metabolische Aufarbeitung unauffällig.
Die Ursache von MAE finden. Das Fehlen erklärender bildgebender oder metabolischer Befunde wirft die Besorgnis über eine zugrunde liegende genetische Ursache auf. Während frühere Studien versucht hatten, einen genetischen Beitrag durch epidemiologische Studien durch Bewertung der Familiengeschichte zu ermitteln, wurde diese traditionelle Methode weitgehend durch neuere Entdeckungen in der Epilepsiegenetik ersetzt. Die Beurteilung der genetischen Grundlagen schwerer Epilepsien im Kindesalter erfolgt heute eher durch Molekulargenetik als durch genetische Epidemiologie. Die Beurteilung der genetischen Architektur von MAE war eines der Ziele in unserer Studie von Tang und Mitarbeitern, die in Epilepsia veröffentlicht wurde.
Gibt es eine Krankheit? Bevor ich die genetischen Befunde bei MAE beschreibe, möchte ich eine laufende Debatte auf dem Gebiet der MAE vorstellen. Die anfängliche Beschreibung durch Doose und Mitarbeiter umfasste weitgehend Kinder mit typischer Entwicklung vor Beginn des Anfalls (> 75%), was mit unserer jüngsten Studie (79%) vergleichbar ist. Betrachten wir jedoch beim Vergleich von Kindern mit und ohne vorherige Entwicklungsprobleme dieselbe Krankheit oder sollten nur Kinder mit vorheriger unauffälliger Entwicklung als typische MAE angesehen werden? Während diese Frage nicht leicht zu lösen ist, wenn man die Epilepsie-Geschichte und die Ergebnisse betrachtet, können die zugrunde liegenden genetischen Veränderungen einige Hinweise geben. Genetische Ursachen werden häufiger bei Kindern mit schwereren neurologischen Entwicklungsstörungen identifiziert und alle Kinder mit identifizierten genetischen Ursachen hatten neben der Epilepsie komplexe neurologische Entwicklungsstörungen. Dementsprechend können genetische Ursachen bei Kindern mit MAE beobachtet werden, typischerweise jedoch nur bei Kindern mit MAE und komorbiden neurologischen Entwicklungsstörungen, was darauf hindeutet, dass MAE nicht nur eine genetisch heterogene Erkrankung ist, sondern ähnliche Merkmale aufweist wie andere Epilepsie im Kindesalter in Bezug auf ihre genetische Heterogenität und Komorbidität. Beispielsweise werden beim West-Syndrom (infantile Spasmen) genetische Ursachen auch nur bei Kindern mit neurologischen Entwicklungsbefunden identifiziert und nicht bei Kindern, die direkt auf Medikamente ohne neurologische Entwicklungsfolgen ansprechen.
Genetische Befunde bei MAE. Das genetische Spektrum bei MAE ist anders als bei den epileptischen Enzephalopathien insgesamt. Gentests wurden bei 85 Personen durchgeführt und 12/85 (14%) Personen hatten positive genetische Befunde. Drei Gene wurden in zwei Individuen gefunden, darunter SYNGAP1, NEXMIF (KIAA2022) und SLC6A1. Diese Triade von Genen wurde zuvor mit Entwicklungs- und epileptischen Enzephalopathien mit generalisierten Anfällen in Verbindung gebracht. Darüber hinaus wurden einzelne Individuen mit krankheitsverursachenden Varianten in KCNA2, SCN2A, STX1B, KCNB1 und MECP2 gefunden. Alle diese Gene wurden zuvor in unserem Blog diskutiert und es ist bekannt, dass sie generalisierte Anfälle und generalisierte EEG-Merkmale aufweisen.
Fehlende Gene in MAE. Während einige frühere Berichte Varianten in SCN1A gefunden haben, wurde dies in unserer Kohorte nicht gefunden. Darüber hinaus fehlen andere häufige Epilepsie-Gene, einschließlich CDKL5 und SLC2A1 (GLUT1). Dies deutet darauf hin, dass die genetische Landschaft von MAE etwas anders ist und sich von anderen Entwicklungs- und epileptischen Enzephalopathien unterscheidet, obwohl nur begrenzte Schlussfolgerungen aus relativ kleinen Kohorten gezogen werden können. Die diagnostische Ausbeute bei MAE ist nicht sehr hoch – 14% ist viel niedriger als typischerweise bei den Entwicklungs- und epileptischen Enzephalopathien. Wenn jedoch Personen mit typischer Entwicklung ausgeschlossen werden, sind die diagnostischen Ausbeuten viel höher, was darauf hindeutet, dass Gentests bei Kindern mit MAE von Wert sind. Obwohl kein einzelnes Individuum mit MAE krankheitsverursachende Varianten in SLC2A1 aufwies, können Gentests auf behandelbare Ursachen genetischer Epilepsien im Allgemeinen bei atypischen generalisierten Epilepsien in Betracht gezogen werden.
Die fehlende Erblichkeit von MAE. Wenn Gentests bei mehr als 80% der Personen negativ auf MAE sind, wo liegt die versteckte genetische Belastung? Ab 2020 würde ich zwei mögliche Erklärungen vorschlagen. Erstens ist die genetische Belastung generalisierter Epilepsien nicht völlig unbekannt. Mehr als 30% des Bevölkerungsrisikos für Erkrankungen wie Abwesenheitsepilepsie im Kindesalter (CAE) oder juvenile myoklonische Epilepsie (JME) werden erklärt. Diese Erklärung wird jedoch nicht durch Einzelgenursachen gegeben, sondern durch ein polygenes Risiko, das sich auf die additive Wirkung Tausender gängiger genetischer Varianten bezieht. Es könnte die Hypothese aufgestellt werden, dass MAE einen „extremen Phänotyp“ generalisierter Epilepsien darstellt, bei denen dieses polygene Risiko besonders hoch ist. Diese Hypothese ist überprüfbar und wird in Zukunft definitiv verfolgt. Darüber hinaus gibt es mehr zum menschlichen Genom als das Exom – neuere Erkenntnisse in den Epilepsien haben die Rolle von nicht-kodierenden Varianten betont, wie Wiederholungsexpansionen bei familiärer erwachsener myoklonischer Epilepsie (FAME). Angesichts der auffallend ähnlichen klinischen Merkmale bei vielen Kindern mit MAE, Es kann immer noch sinnvoll sein, für viele Kinder mit MAE außerhalb des Exoms nach einer gemeinsamen genetischen Ursache zu suchen, Verwendung der Sequenzierung des gesamten Genoms. Zusammengenommen sind die Bewertung polygener Risikoscores (PRS) und die Sequenzierung des gesamten Genoms (WGS) zwei mögliche Wege, um die genetischen Grundlagen von MAE weiter zu identifizieren.
Was Sie wissen müssen. Das Doose-Syndrom (MAE) ist ein vermutlich genetisches Epilepsiesyndrom mit heterogener Ursache. Krankheitsverursachende Varianten sind bei 14% der Kinder vorhanden und werden typischerweise bei Kindern mit zusätzlichen neurologischen Entwicklungsmerkmalen wie Entwicklungsverzögerung oder Autismus identifiziert. Eine Triade von Genen, einschließlich SYNGAP1, NEXMIF (KIAA2022) und SLC6A1, sind wiederkehrende genetische Ursachen, die auf eine gemeinsame zugrunde liegende Biologie hinweisen können, die sich etwas von anderen Entwicklungs- und epileptischen Enzephalopathien unterscheidet.

Ingo Helbig ist Kinderneurologe und Epilepsie-Genetiker am Children’s Hospital of Philadelphia (CHOP), USA. Er leitet auch die Epilepsie-Genetik-Gruppe an der Universität Kiel.

