MAE. Hay muchos síndromes de epilepsia infantil distintos de los que nos hemos dado cuenta críticamente en la era genómica, ya que están vinculados a causas genéticas prominentes, incluido el Síndrome de Dravet (SCN1A) y la Epilepsia de la Infancia con Convulsiones Focales Migratorias (KCNT1). Sin embargo, hay muchos otros síndromes de epilepsia en los que se ha sospechado durante mucho tiempo una causa genética, pero que ha permanecido esquiva. Uno de los síndromes de epilepsia que en gran medida ha permanecido inexplorado es el Síndrome de Doose, también conocido como Epilepsia Astática Mioclónica (MAE) o Epilepsia con Convulsiones Mioclónicas-Atónicas. En un estudio reciente en Epilepsia, exploramos la arquitectura genética del Síndrome de Doose e identificamos causas monogénicas en el 14% de los individuos, incluidos SYNGAP1, NEXMIF (KIAA2022) y SLC6A1. Nuestro estudio sugiere que el Síndrome de Doose es genéticamente heterogéneo, posiblemente con un paisaje genético distinto.
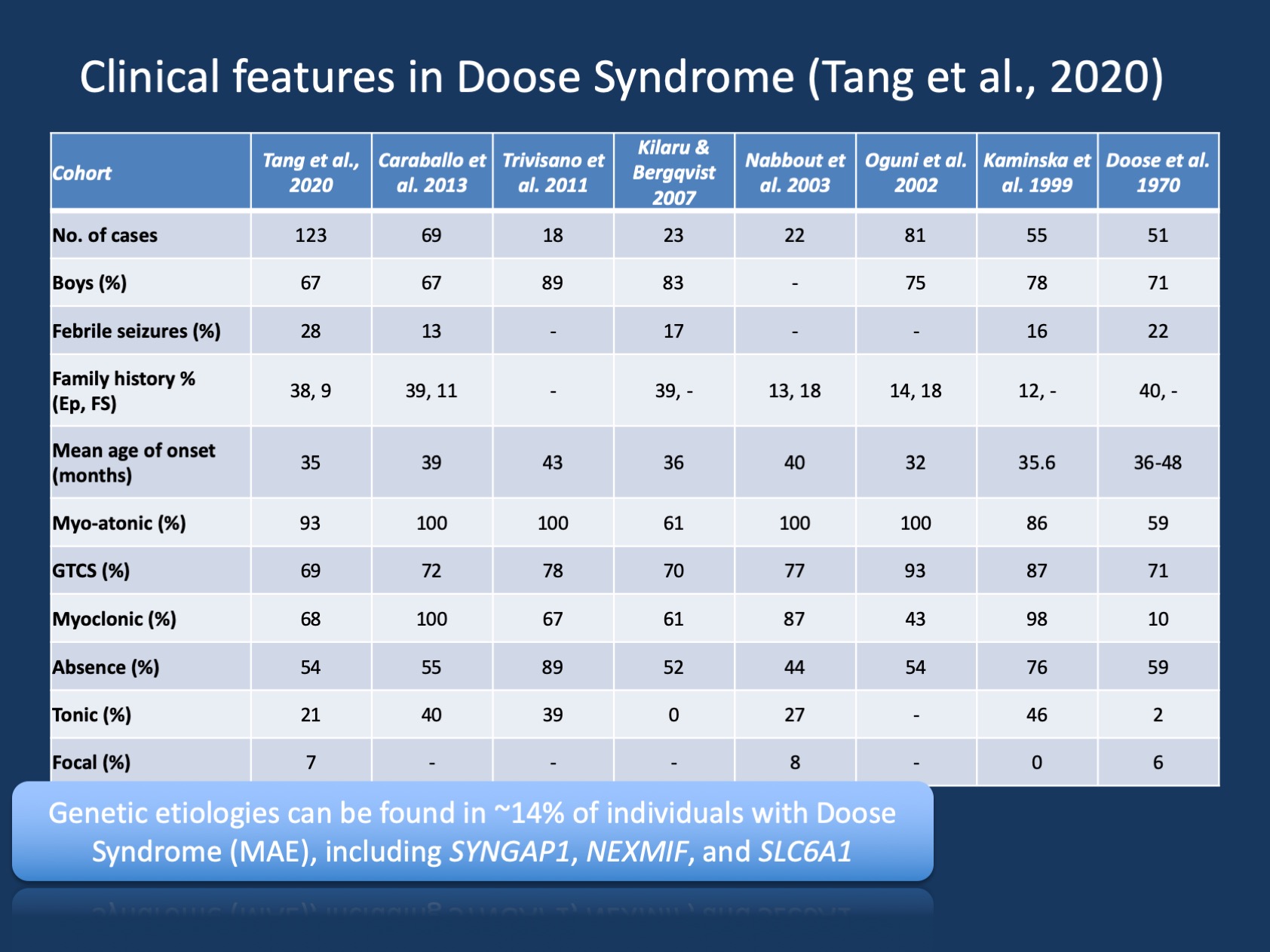
Gráfico 1 Características clínicas del Síndrome de Doose (epilepsia con convulsiones atónicas mioclónicas o Epilepsia Astática Mioclónica ). La tabla es una adaptación de nuestra reciente publicación de Tang et al., 2020 y compara las características clínicas en las grandes cohortes de Síndrome de Doose que se han notificado desde la descripción inicial en 1970. El síndrome de Doose (MAE) es un síndrome de epilepsia poco frecuente, pero es una entidad clínica reconocible. Sin embargo, en contraste con el Síndrome de Dravet, en el que >90% de los individuos tienen variantes causantes de enfermedades en SCN1A, el Síndrome de Doose es genéticamente heterogéneo.
La historia de MAE. En primer lugar, permítanme aclarar algunas cuestiones de nombres. El Síndrome de Doose o Epilepsia Astática Mioclónica (MAE) ahora se conoce oficialmente como Epilepsia con Convulsiones Mioclónicas-Atónicas utilizando la convención de nomenclatura más reciente. Sin embargo, aunque normalmente tratamos de alinearnos con la forma oficial de cómo se usan los términos relacionados con la epilepsia, permítanme ser un poco terco aquí. Como neurólogo infantil entrenado en el Departamento de Neuropediatría en Kiel, Alemania, me referiré a esta afección como Síndrome de Doose usando MAE como abreviatura, en honor a Hermann Doose (1927-2018), uno de los fundadores de la epileptología pediátrica europea (enlace a ILAE in memoriam). Muchos hablantes nativos de inglés suelen equivocarse en la pronunciación: la pronunciación correcta es «Dohs-ah» y NO rima con «goose».
El cuadro clínico de MAE. Doose se acredita típicamente con hacer una observación clínica importante: en la década de 1960, él y sus colaboradores en Kiel reconocieron una epilepsia inusual en niños que se caracterizaba por el inicio repentino de ataques de caída (convulsiones atónicas), convulsiones mioclónicas, convulsiones tónico-clónicas generalizadas y un patrón de EEG generalizado, que ocurre típicamente entre los dos y los cinco años de edad. Esta epilepsia se presentó típicamente en niños con un desarrollo típico previo (ver a continuación) y a menudo resultó en retraso del desarrollo y discapacidad intelectual. Doose y colaboradores reconocieron que este síndrome de epilepsia recién reconocido era diferente del Síndrome de Lennox-Gastaut o las epilepsias con convulsiones tónico-clónicas generalizadas precipitadas por fiebre, las últimas de las cuales se conceptualizaron posteriormente como Síndrome de Dravet.
Entendiendo MAE. Las descripciones clínicas del Síndrome de Doose (MAE) han sido constantes a lo largo de los años, pero MAE no ha recibido tanta atención como los síndromes de epilepsia infantil en los que se había descubierto una base genética y en los que la investigación dirigida puede permitir tratamientos nuevos. En nuestra reciente publicación de Tang y colaboradores, hemos enumerado ocho estudios en los últimos 50 años entre 1970 y 2020 con un total de 442 individuos descritos. En consecuencia, la EMA es poco frecuente, pero es lo suficientemente común como para que la mayoría de los neurólogos infantiles y epileptólogos pediátricos estén al tanto de esta afección. Solo un pequeño subgrupo de personas con EAM tiene hallazgos estructurales en el cerebro o anomalías metabólicas que explican la epilepsia subyacente; en la gran mayoría de los niños con EAM, la neuroimagen y el análisis metabólico no son notables.
Encontrar la causa de MAE. La ausencia de imágenes explicativas o hallazgos metabólicos plantea la preocupación por una causa genética subyacente. Si bien los estudios anteriores habían tratado de establecer una contribución genética a través de estudios epidemiológicos mediante la evaluación de las historias familiares, este método tradicional ha sido reemplazado en gran medida por los recientes descubrimientos en genética de la epilepsia. La evaluación de la base genética de las epilepsias infantiles graves se realiza ahora a través de la genética molecular en lugar de la epidemiología genética. Evaluar la arquitectura genética de MAE fue uno de los objetivos de nuestro estudio de Tang y colaboradores que se publicó en Epilepsia.
¿Es MAE una enfermedad? Antes de describir los hallazgos genéticos en MAE, permítanme presentar un debate en curso dentro del campo de MAE. La descripción inicial de Doose y colaboradores incluyó en gran medida a niños con un desarrollo típico antes de la aparición de las convulsiones (>75%), lo que es comparable a nuestro estudio reciente (79%). Sin embargo, cuando se comparan niños con y sin problemas de desarrollo previos, ¿estamos mirando la misma enfermedad o solo se debe considerar a los niños con un desarrollo normal previo como el típico EAM? Si bien esta pregunta no se resuelve fácilmente al considerar los antecedentes y los resultados de la epilepsia, las alteraciones genéticas subyacentes pueden proporcionar algunas pistas. Las causas genéticas se identifican con mayor frecuencia en niños con trastornos del neurodesarrollo más graves y todos los niños con causas genéticas identificadas tenían trastornos complejos del neurodesarrollo además de la epilepsia. En consecuencia, se pueden observar causas genéticas en niños con EAM, pero por lo general solo en niños con EAM y trastornos comórbidos del neurodesarrollo, lo que sugiere que la EAM no es solo una afección genéticamente heterogénea, sino que tiene características similares a otras epilepsia en la infancia con respecto a su heterogeneidad genética y comorbilidad. Por ejemplo, en el Síndrome de West (Espasmos Infantiles), las causas genéticas también se identifican solo en niños con hallazgos en el desarrollo neurológico y no en niños que responden directamente a medicamentos sin secuelas en el desarrollo neurológico.
Hallazgos genéticos en EAM. El espectro genético en MAE es diferente al de las encefalopatías epilépticas en general. Se realizaron pruebas genéticas en 85 individuos y 12/85 (14%) individuos tuvieron hallazgos genéticos positivos. Se encontraron tres genes en dos individuos, incluyendo SYNGAP1, NEXMIF (KIAA2022) y SLC6A1. Esta tríada de genes se ha relacionado previamente con encefalopatías epilépticas y de desarrollo con convulsiones generalizadas. Además, se encontraron individuos únicos con variantes causantes de enfermedad en KCNA2, SCN2A, STX1B, KCNB1 y MECP2. Todos estos genes fueron discutidos previamente en nuestro blog y se sabe que se presentan con convulsiones generalizadas y características de EEG generalizadas.
Genes ausentes en MAE. Si bien algunos informes anteriores han encontrado variantes en el SCN1A, esto no se encontró en nuestra cohorte. Además, otros genes comunes de epilepsia están notablemente ausentes, incluidos CDKL5 y SLC2A1 (GLUT1). Esto sugiere que el paisaje genético de MAE es algo distinto y diferente de otras encefalopatías epilépticas y de desarrollo, a pesar de que solo se pueden extraer conclusiones limitadas de cohortes relativamente pequeñas. El rendimiento diagnóstico en EMA no es muy alto: el 14% es mucho menor que el que se observa normalmente en las encefalopatías epilépticas y de desarrollo. Sin embargo, al excluir a individuos con desarrollo típico, los rendimientos diagnósticos son mucho más altos, lo que sugiere que las pruebas genéticas son valiosas en niños con EAM. En general, a pesar de que ningún individuo con EMA tenía variantes causantes de enfermedad en SLC2A1, las pruebas genéticas para las causas tratables de epilepsias genéticas generalmente se pueden considerar en las epilepsias generalizadas atípicas.
La falta de heredabilidad de MAE. Si las pruebas genéticas son negativas para MAE en más del 80% de los individuos, ¿dónde está la carga genética oculta? A partir de 2020, sugeriría dos posibles explicaciones. En primer lugar, la carga genética de las epilepsias generalizadas no es completamente desconocida. Más del 30% de la población tiene riesgo de padecer afecciones como la Epilepsia de Ausencia Infantil (CAE) o la Epilepsia Mioclónica Juvenil (JME). Sin embargo, esta explicación no se da por causas de un solo gen, sino por el riesgo poligénico, refiriéndose al efecto aditivo de miles de variantes genéticas comunes. Se podría plantear la hipótesis de que MAE puede representar un «fenotipo extremo» de epilepsias generalizadas donde este riesgo poligénico es particularmente alto. Esta hipótesis es comprobable, y sin duda continuará en el futuro. Además, hay más en el genoma humano que en el exoma: los hallazgos recientes en las epilepsias han enfatizado el papel de las variantes no codificantes, como las expansiones repetidas en la Epilepsia Mioclónica Familiar Adulta (FAME). Dadas las características clínicas sorprendentemente similares en muchos niños con EMA, todavía puede ser razonable buscar una causa genética común para muchos niños con EMA fuera del exoma, utilizando la secuenciación del genoma completo. En conjunto, la evaluación de las puntuaciones de riesgo poligénico (PRS, por sus siglas en inglés) y la secuenciación del genoma completo (WGS, por sus siglas en inglés) son dos vías potenciales para identificar aún más la base genética de MAE.
Lo que necesita saber. El síndrome de Doose (MAE) es un síndrome epiléptico genético con una causa heterogénea. Las variantes causantes de enfermedades están presentes en el 14% de los niños y, por lo general, se identifican en niños con características adicionales del neurodesarrollo, como retraso del desarrollo o autismo. Una tríada de genes que incluyen SYNGAP1, NEXMIF (KIAA2022) y SLC6A1, son causas genéticas recurrentes que pueden sugerir una biología subyacente compartida que es algo distinta de otras encefalopatías epilépticas y de desarrollo.

Ingo Helbig es neuróloga infantil e investigadora de genética de epilepsia que trabaja en el Hospital Infantil de Filadelfia (CHOP), EE. También dirige el grupo de genética de la epilepsia en la Universidad de Kiel, Alemania.

