MAE. Il existe de nombreux syndromes d’épilepsie infantile distincts dont nous avons pris conscience de manière critique à l’ère génomique car ils sont liés à des causes génétiques importantes, notamment le syndrome de Dravet (SCN1A) et l’épilepsie de la petite enfance avec crises focales migratrices (KCNT1). Cependant, il existe de nombreux autres syndromes d’épilepsie où une cause génétique a longtemps été suspectée, mais est restée insaisissable. L’un des syndromes d’épilepsie qui est largement resté inexploré est le syndrome de Doose, également appelé Épilepsie Astatique Myoclonique (EMA) ou Épilepsie avec crises Myocloniques-Atoniques. Dans une étude récente sur l’épilepsie, nous avons exploré l’architecture génétique du syndrome de Doose et identifié des causes monogènes chez 14% des individus, y compris SYNGAP1, NEXMIF (KIAA2022) et SLC6A1. Notre étude suggère que le syndrome de Doose est génétiquement hétérogène, éventuellement avec un paysage génétique distinct.
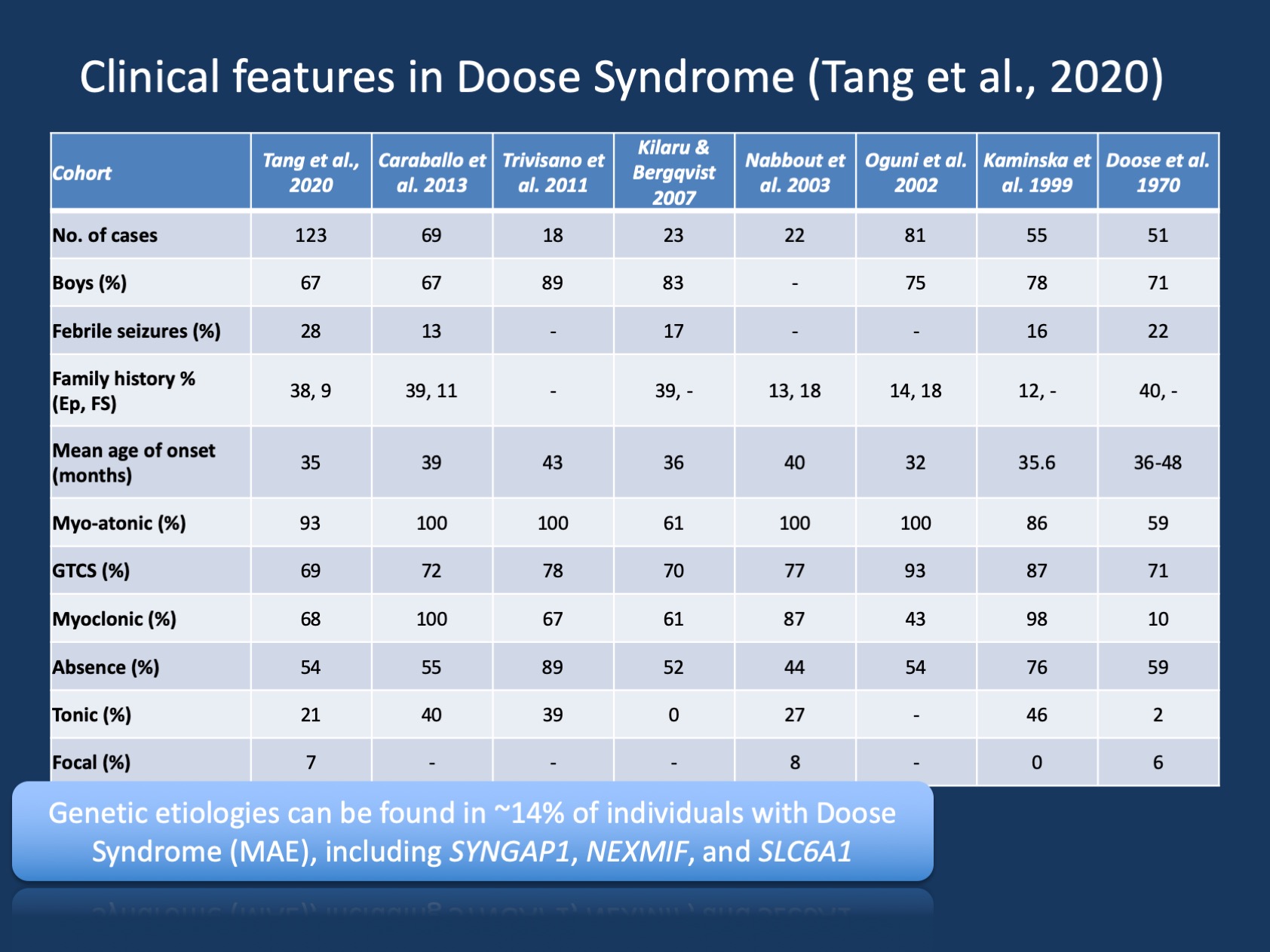
Figure 1. Les caractéristiques cliniques du syndrome de Doose (épilepsie avec crises atoniques myocloniques ou Épilepsie Astatique myoclonique). Le tableau est adapté de notre récente publication de Tang et al., 2020 et compare les caractéristiques cliniques dans les grandes cohortes de syndrome de Doose qui ont été rapportées depuis la description initiale en 1970. Le syndrome de Doose (MAE) est un syndrome d’épilepsie rare, mais constitue une entité clinique reconnaissable. Cependant, contrairement au syndrome de Dravet où > 90% des individus présentent des variantes pathogènes du SCN1A, le syndrome de Doose est génétiquement hétérogène.
L’histoire de MAE. Tout d’abord, permettez-moi d’éclaircir certains problèmes de nommage. Le syndrome de Doose ou Épilepsie Astatique myoclonique (EMA) est maintenant officiellement appelé Épilepsie avec Crises Myocloniques-Atoniques en utilisant la convention de dénomination la plus récente. Cependant, alors que nous essayons généralement de nous aligner sur la manière officielle d’utiliser les termes liés à l’épilepsie, permettez-moi d’être un peu têtu ici. En tant que neurologue de l’enfant formé au Département de Neuropédiatrie de Kiel, en Allemagne, je qualifierai cette affection de Syndrome de Doose en utilisant MAE comme raccourci, en hommage à Hermann Doose (1927-2018) qui est l’un des fondateurs de l’épileptologie pédiatrique européenne (lien vers ILAE in memoriam). De nombreux anglophones natifs se trompent généralement de prononciation – la prononciation correcte est « Dohs-ah » et ne rime PAS avec « goose ».
Le tableau clinique de la MAE. Doose est généralement crédité d’avoir fait une observation clinique importante: dans les années 1960, lui et ses collaborateurs à Kiel ont reconnu une épilepsie inhabituelle chez les enfants qui se caractérisait par l’apparition soudaine d’attaques de goutte (crises atoniques), de crises myocloniques, de crises tonico-cloniques généralisées et d’un schéma EEG généralisé, survenant généralement entre l’âge de deux et cinq ans. Cette épilepsie se produisait généralement chez des enfants ayant un développement typique antérieur (voir ci-dessous) et entraînait souvent un retard de développement et une déficience intellectuelle. Doose et ses collaborateurs ont reconnu que ce syndrome d’épilepsie nouvellement reconnu était différent du syndrome de Lennox-Gastaut ou des épilepsies avec crises tonico-cloniques généralisées précipitées par la fièvre, ces dernières ayant ensuite été conceptualisées sous le nom de syndrome de Dravet.
Comprendre MAE. Les descriptions cliniques du syndrome de Doose (EMA) ont été stables au fil des ans, mais l’EMA n’a pas reçu autant d’attention que les syndromes d’épilepsie infantile où une base génétique avait été découverte et où des recherches ciblées pourraient permettre de nouveaux traitements. Dans notre récente publication par Tang et ses collaborateurs, nous avons répertorié huit études au cours des 50 dernières années entre 1970 et 2020 avec un total de 442 individus décrits. En conséquence, la MAE est rare, mais elle est assez courante pour que la plupart des neurologues de l’enfant et des épileptologues pédiatriques soient conscients de cette condition. Seul un petit sous-ensemble d’individus atteints d’EMA présente des découvertes cérébrales structurelles ou des anomalies métaboliques qui expliquent l’épilepsie sous–jacente – chez la grande majorité des enfants atteints d’EMA, la neuroimagerie et le travail métabolique ne sont pas remarquables.
Trouver la cause de l’EMA. L’absence d’imagerie explicative ou de résultats métaboliques soulève la préoccupation d’une cause génétique sous-jacente. Alors que des études antérieures avaient tenté de mettre en évidence une contribution génétique par le biais d’études épidémiologiques en évaluant les antécédents familiaux, cette méthode traditionnelle a été largement remplacée par des découvertes récentes en génétique de l’épilepsie. L’évaluation de la base génétique des épilepsies infantiles sévères se fait maintenant par la génétique moléculaire plutôt que par l’épidémiologie génétique. L’évaluation de l’architecture génétique de la MAE était l’un des objectifs de notre étude menée par Tang et ses collaborateurs et publiée dans Epilepsia.
La MAE est-elle une maladie? Avant de décrire les découvertes génétiques dans l’EMA, permettez-moi d’introduire un débat en cours dans le domaine de l’EMA. La description initiale par Doose et ses collaborateurs comprenait en grande partie des enfants ayant un développement typique avant le début de la crise (> 75%), ce qui est comparable à notre étude récente (79%). Cependant, lorsque l’on compare des enfants avec et sans problèmes de développement antérieurs, examinons-nous la même maladie ou seuls les enfants ayant un développement banal antérieur devraient-ils être considérés comme des MAE typiques? Bien que cette question ne soit pas facilement résolue lorsque l’on considère les antécédents et les résultats de l’épilepsie, les altérations génétiques sous-jacentes peuvent fournir des indices. Les causes génétiques sont identifiées plus fréquemment chez les enfants présentant des troubles neurodéveloppementaux plus graves et tous les enfants présentant des causes génétiques identifiées présentaient des troubles neurodéveloppementaux complexes en plus de l’épilepsie. En conséquence, des causes génétiques peuvent être observées chez les enfants atteints d’EMA, mais généralement uniquement chez les enfants atteints d’EMA et de troubles neurodéveloppementaux comorbides, suggérant que l’EMA n’est pas seulement une maladie génétiquement hétérogène, mais présente des caractéristiques similaires à celles des autres épilepsies de l’enfance en ce qui concerne leur hétérogénéité génétique et leur comorbidité. Par exemple, dans le syndrome de West (Spasmes infantiles), les causes génétiques ne sont également identifiées que chez les enfants présentant des résultats neurodéveloppementaux et non chez les enfants qui répondent directement aux médicaments sans séquelles neurodéveloppementales.
Résultats génétiques dans l’EMM. Le spectre génétique de l’EMA est différent de celui des encéphalopathies épileptiques en général. Des tests génétiques ont été effectués chez 85 personnes et 12 sur 85 (14 %) avaient des résultats génétiques positifs. Trois gènes ont été trouvés chez deux individus, dont SYNGAP1, NEXMIF (KIAA2022) et SLC6A1. Cette triade de gènes a déjà été liée à des encéphalopathies développementales et épileptiques avec crises généralisées. De plus, des individus isolés ont été trouvés avec des variantes pathogènes dans KCNA2, SCN2A, STX1B, KCNB1 et MECP2. Tous ces gènes ont déjà été discutés sur notre blog et sont connus pour présenter des crises généralisées et des caractéristiques EEG généralisées.
Gènes absents dans l’EMA. Bien que certains rapports antérieurs aient trouvé des variantes dans SCN1A, cela n’a pas été trouvé dans notre cohorte. De plus, d’autres gènes courants d’épilepsie sont notablement absents, notamment CDKL5 et SLC2A1 (GLUT1). Cela suggère que le paysage génétique de l’EMA est quelque peu distinct et différent des autres encéphalopathies développementales et épileptiques, même si seules des conclusions limitées peuvent être tirées à partir de cohortes relativement petites. Le rendement diagnostique dans l’EMA n’est pas très élevé – 14% est beaucoup plus faible que ce qui est généralement observé dans les encéphalopathies développementales et épileptiques. Cependant, en excluant les individus ayant un développement typique, les rendements diagnostiques sont beaucoup plus élevés, ce qui suggère que les tests génétiques sont utiles chez les enfants atteints d’EMA. En général, même si aucun individu atteint d’EMA ne présentait de variants pathogènes dans le SLC2A1, les tests génétiques pour détecter les causes traitables des épilepsies génétiques peuvent généralement être envisagés dans les épilepsies généralisées atypiques.
L’héritabilité manquante de MAE. Si les tests génétiques sont négatifs pour l’EMA chez plus de 80% des individus, où est le fardeau génétique caché? À partir de 2020, je suggérerais deux explications possibles. Premièrement, le fardeau génétique des épilepsies généralisées n’est pas entièrement inconnu. Plus de 30% du risque de maladies telles que l’Épilepsie d’absence infantile (IAO) ou l’Épilepsie myoclonique juvénile (EMJ) est expliqué. Cependant, cette explication n’est pas donnée par des causes monogéniques, mais par un risque polygénique, se référant à l’effet additif de milliers de variantes génétiques courantes. On pourrait émettre l’hypothèse que l’EMA pourrait représenter un « phénotype extrême » d’épilepsies généralisées où ce risque polygénique est particulièrement élevé. Cette hypothèse est testable et sera certainement poursuivie à l’avenir. De plus, il y a plus dans le génome humain que l’exome – des résultats récents dans les épilepsies ont souligné le rôle des variantes non codantes, telles que les expansions répétées dans l’épilepsie myoclonique familiale adulte (FAME). Étant donné les caractéristiques cliniques étonnamment similaires chez de nombreux enfants atteints d’EMA, il peut toujours être raisonnable de rechercher une cause génétique commune pour de nombreux enfants atteints d’EMA en dehors de l’exome, en utilisant le séquençage du génome entier. Dans l’ensemble, l’évaluation des scores de risque polygénique (PRS) et du séquençage du génome entier (WGS) sont deux pistes potentielles pour identifier davantage la base génétique de l’EMA.
Ce que vous devez savoir. Le syndrome de Doose (MAE) est un syndrome d’épilepsie vraisemblablement génétique de cause hétérogène. Des variantes pathogènes sont présentes chez 14% des enfants et sont généralement identifiées chez les enfants présentant des caractéristiques neurodéveloppementales supplémentaires telles que le retard de développement ou l’autisme. Une triade de gènes, dont SYNGAP1, NEXMIF (KIAA2022) et SLC6A1, sont des causes génétiques récurrentes qui peuvent faire allusion à une biologie sous-jacente partagée quelque peu distincte des autres encéphalopathies développementales et épileptiques.

Ingo Helbig est neurologue pour enfants et chercheur en génétique de l’épilepsie travaillant à l’Hôpital pour enfants de Philadelphie (CHOP), aux États-Unis. Il dirige également le groupe de génétique de l’épilepsie à l’Université de Kiel, en Allemagne.

