- Milieu des années 1800 – 1945: Des plantes médicinales aux premières drogues de synthèsmodifier
- Épinéphrine, noradrénaline et amphétamineSdit
- Découverte et développement des barbituriquesmodifier
- InsulinEdit
- Recherche anti-infectieuse précoce: Salvarsan, Prontosil, Pénicilline et vaccinesmodifier
- Médicaments dangereux et réglementation précoce de l’industriemodifier
- Les années d’après-guerre, 1945– 1970modifier
- De nouveaux progrès dans la recherche anti-infectieuxmodifier
- Développement et commercialisation de médicaments antihypertenseurs
- Contraceptifs orauxmodifier
- Thalidomide et les amendements Kefauver-Harris.
- 1970–1980sEdit
- Statinesedit
Milieu des années 1800 – 1945: Des plantes médicinales aux premières drogues de synthèsmodifier
L’industrie pharmaceutique moderne a commencé avec les apothicaires locaux qui sont passés de leur rôle traditionnel de distribution de médicaments botaniques tels que la morphine et la quinine à la fabrication en gros au milieu des années 1800, et à partir des découvertes résultant de la recherche appliquée. La découverte intentionnelle de médicaments à partir de plantes a commencé avec l’isolement entre 1803 et 1805 de la morphine – un analgésique et un agent induisant le sommeil – de l’opium par l’assistant apothicaire allemand Friedrich Sertürner, qui a nommé ce composé d’après le dieu grec des rêves, Morphée. À la fin des années 1880, les fabricants de colorants allemands avaient perfectionné la purification de composés organiques individuels à partir de goudron et d’autres sources minérales et avaient également mis en place des méthodes rudimentaires de synthèse chimique organique. Le développement de méthodes chimiques de synthèse a permis aux scientifiques de varier systématiquement la structure des substances chimiques, et la croissance de la science émergente de la pharmacologie a élargi leur capacité à évaluer les effets biologiques de ces changements structurels.
Épinéphrine, noradrénaline et amphétamineSdit
Dans les années 1890, l’effet profond des extraits surrénaliens sur de nombreux types de tissus différents avait été découvert, déclenchant une recherche à la fois du mécanisme de signalisation chimique et des efforts pour exploiter ces observations pour le développement de nouveaux médicaments. L’augmentation de la pression artérielle et les effets vasoconstricteurs des extraits surrénaliens intéressaient particulièrement les chirurgiens en tant qu’agents hémostatiques et en tant que traitement du choc, et un certain nombre de sociétés ont développé des produits à base d’extraits surrénaliens contenant des puretés variables de la substance active. En 1897, John Abel de l’Université Johns Hopkins a identifié le principe actif comme étant l’épinéphrine, qu’il a isolée à l’état impur sous forme de sel de sulfate. Le chimiste industriel Jōkichi Takamine a ensuite développé une méthode pour obtenir de l’épinéphrine à l’état pur, et a concédé la technologie sous licence à Parke-Davis. Parke-Davis a commercialisé l’épinéphrine sous le nom commercial Adrenalin. L’épinéphrine injectée s’est avérée particulièrement efficace pour le traitement aigu des crises d’asthme, et une version inhalée a été vendue aux États-Unis jusqu’en 2011 (Primatene Mist). En 1929, l’épinéphrine avait été formulée en inhalateur pour le traitement de la congestion nasale.
Bien que très efficace, la nécessité d’une injection limitait l’utilisation de l’épinéphrine et des dérivés actifs par voie orale ont été recherchés. Un composé structurellement similaire, l’éphédrine (en fait plus similaire à la noradrénaline), a été identifié par des chimistes japonais dans l’usine de Ma Huang et commercialisé par Eli Lilly comme traitement oral de l’asthme. À la suite des travaux de Henry Dale et George Barger chez Burroughs-Wellcome, le chimiste universitaire Gordon Alles a synthétisé l’amphétamine et l’a testée chez des patients asthmatiques en 1929. Le médicament s’est avéré n’avoir que des effets anti-asthmatiques modestes, mais a produit des sensations d’exaltation et de palpitations. L’amphétamine a été développée par Smith, Kline et French comme décongestionnant nasal sous le nom commercial Inhalateur de benzédrine. L’amphétamine a finalement été développée pour le traitement de la narcolepsie, du parkinsonisme post-encéphalitique et de l’élévation de l’humeur dans la dépression et d’autres indications psychiatriques. Il a reçu l’approbation comme remède nouveau et Non officiel de l’Association médicale américaine pour ces utilisations en 1937 et est resté d’usage courant pour la dépression jusqu’au développement des antidépresseurs tricycliques dans les années 1960.
Découverte et développement des barbituriquesmodifier

En 1903, Hermann Emil Fischer et Joseph von Mering ont révélé que l’acide diéthylbarbiturique, formé à partir de la réaction de l’acide diéthylmalonique, de l’oxychlorure de phosphore et de l’urée, induit le sommeil chez les chiens. La découverte a été brevetée et concédée sous licence à Bayer pharmaceuticals, qui a commercialisé le composé sous le nom commercial Veronal comme aide au sommeil à partir de 1904. Des études systématiques de l’effet des changements structurels sur la puissance et la durée d’action ont conduit à la découverte du phénobarbital chez Bayer en 1911 et à la découverte de sa puissante activité antiépileptique en 1912. Le phénobarbital était l’un des médicaments les plus utilisés pour le traitement de l’épilepsie dans les années 1970 et, en 2014, il figure sur la liste des médicaments essentiels de l’Organisation mondiale de la santé. Les années 1950 et 1960 ont vu une prise de conscience accrue des propriétés addictives et du potentiel d’abus des barbituriques et des amphétamines et ont conduit à des restrictions croissantes sur leur utilisation et à une surveillance gouvernementale croissante des prescripteurs. Aujourd’hui, l’amphétamine est largement limitée dans le traitement du trouble déficitaire de l’attention et le phénobarbital dans le traitement de l’épilepsie.
InsulinEdit
Une série d’expériences réalisées de la fin des années 1800 au début des années 1900 a révélé que le diabète est causé par l’absence d’une substance normalement produite par le pancréas. En 1869, Oskar Minkowski et Joseph von Mering ont découvert que le diabète pouvait être induit chez le chien par une ablation chirurgicale du pancréas. En 1921, le professeur canadien Frederick Banting et son étudiant Charles Best ont répété cette étude et ont constaté que les injections d’extrait pancréatique inversaient les symptômes produits par l’ablation du pancréas. Bientôt, il a été démontré que l’extrait fonctionnait chez l’homme, mais le développement de l’insulinothérapie en tant qu’intervention médicale de routine a été retardé par des difficultés à produire le matériau en quantité suffisante et avec une pureté reproductible. Les chercheurs ont demandé l’aide de collaborateurs industriels chez Eli Lilly and Co. basé sur l’expérience de l’entreprise avec la purification à grande échelle de matériaux biologiques. Le chimiste George B. Walden d’Eli Lilly and Company a constaté qu’un ajustement minutieux du pH de l’extrait permettait de produire une insuline de qualité relativement pure. Sous la pression de l’Université de Toronto et une contestation possible des brevets par des scientifiques universitaires qui avaient mis au point indépendamment une méthode de purification similaire, un accord a été conclu pour la production non exclusive d’insuline par plusieurs entreprises. Avant la découverte et la disponibilité généralisée de l’insulinothérapie, l’espérance de vie des diabétiques n’était que de quelques mois.
Recherche anti-infectieuse précoce: Salvarsan, Prontosil, Pénicilline et vaccinesmodifier
Le développement de médicaments pour le traitement des maladies infectieuses était l’un des principaux axes des premiers efforts de recherche et de développement; en 1900, la pneumonie, la tuberculose et la diarrhée étaient les trois principales causes de décès aux États-Unis et la mortalité au cours de la première année de vie dépassait 10%.
En 1911, l’arsphénamine, le premier médicament anti-infectieux synthétique, a été mis au point par Paul Ehrlich et le chimiste Alfred Bertheim de l’Institut de thérapie expérimentale de Berlin. Le médicament a reçu le nom commercial Salvarsan. Ehrlich, notant à la fois la toxicité générale de l’arsenic et l’absorption sélective de certains colorants par les bactéries, a émis l’hypothèse qu’un colorant contenant de l’arsenic ayant des propriétés d’absorption sélective similaires pourrait être utilisé pour traiter les infections bactériennes. L’arsphénamine a été préparée dans le cadre d’une campagne visant à synthétiser une série de ces composés et s’est avérée présenter une toxicité partiellement sélective. L’arsphénamine s’est avérée être le premier traitement efficace de la syphilis, une maladie qui, auparavant, était incurable et entraînait inexorablement de graves ulcérations cutanées, des lésions neurologiques et la mort.
L’approche d’Ehrlich consistant à faire varier systématiquement la structure chimique des composés synthétiques et à mesurer les effets de ces changements sur l’activité biologique a été largement poursuivie par les scientifiques industriels, notamment les scientifiques de Bayer Josef Klarer, Fritz Mietzsch et Gerhard Domagk. Ce travail, également basé sur le test de composés disponibles dans l’industrie des colorants allemande, a conduit au développement du Prontosil, le premier représentant de la classe des antibiotiques sulfamides. Comparés à l’arsphénamine, les sulfamides avaient un spectre d’activité plus large et étaient beaucoup moins toxiques, les rendant utiles pour les infections causées par des agents pathogènes tels que les streptocoques. En 1939, Domagk a reçu le prix Nobel de médecine pour cette découverte. Néanmoins, la diminution spectaculaire des décès dus aux maladies infectieuses qui a eu lieu avant la Seconde Guerre mondiale était principalement le résultat de l’amélioration des mesures de santé publique telles que l’eau potable et des logements moins surpeuplés, et l’impact des médicaments et des vaccins anti-infectieux a été important principalement après la Seconde Guerre mondiale.
En 1928, Alexander Fleming découvrit les effets antibactériens de la pénicilline, mais son exploitation pour le traitement des maladies humaines attendait le développement de méthodes pour sa production et sa purification à grande échelle. Ceux-ci ont été développés par un consortium de sociétés pharmaceutiques dirigé par les gouvernements américain et britannique pendant la Seconde Guerre mondiale.
Les premiers progrès vers la mise au point de vaccins se sont produits tout au long de cette période, principalement sous la forme de recherches fondamentales universitaires et financées par le gouvernement visant à identifier les agents pathogènes responsables des maladies transmissibles courantes. En 1885, Louis Pasteur et Pierre Paul Émile Roux créent le premier vaccin antirabique. Les premiers vaccins contre la diphtérie ont été produits en 1914 à partir d’un mélange de toxine diphtérique et d’antitoxine (produite à partir du sérum d’un animal inoculé), mais l’innocuité de l’inoculation était marginale et elle n’était pas largement utilisée. Les États-Unis ont enregistré 206 000 cas de diphtérie en 1921, entraînant 15 520 décès. En 1923, des efforts parallèles de Gaston Ramon à l’Institut Pasteur et d’Alexander Glenny aux Laboratoires de recherche Wellcome (qui feront plus tard partie de GlaxoSmithKline) ont conduit à la découverte qu’un vaccin plus sûr pourrait être produit en traitant la toxine diphtérique avec du formaldéhyde. En 1944, Maurice Hilleman de Squibb Pharmaceuticals a développé le premier vaccin contre l’encéphélite japonaise. Hilleman déménagera plus tard chez Merck où il jouera un rôle clé dans le développement de vaccins contre la rougeole, les oreillons, la varicelle, la rubéole, l’hépatite A, l’hépatite B et la méningite.
Médicaments dangereux et réglementation précoce de l’industriemodifier

Avant le 20e siècle, les médicaments étaient généralement produits par des fabricants à petite échelle avec peu de contrôle réglementaire sur la fabrication ou les allégations d’innocuité et d’efficacité. Dans la mesure où de telles lois existaient, leur application était laxiste. Aux États-Unis, la réglementation accrue des vaccins et autres médicaments biologiques a été stimulée par des épidémies de tétanos et des décès causés par la distribution de vaccins contaminés contre la variole et d’antitoxine diphtérique. La Loi sur le contrôle des produits biologiques de 1902 exigeait que le gouvernement fédéral accorde une approbation préalable à la mise en marché pour chaque médicament biologique et pour le procédé et l’installation produisant de tels médicaments. Cela a été suivi en 1906 par la Pure Food and Drugs Act, qui interdisait la distribution interétatique d’aliments et de drogues frelatés ou mal étiquetés. Une drogue était considérée comme mal étiquetée si elle contenait de l’alcool, de la morphine, de l’opium, de la cocaïne ou l’une de plusieurs autres drogues potentiellement dangereuses ou addictives, et si son étiquette n’indiquait pas la quantité ou la proportion de ces drogues. Les tentatives du gouvernement d’utiliser la loi pour poursuivre les fabricants pour avoir fait des allégations d’efficacité non étayées ont été sapées par une décision de la Cour suprême limitant les pouvoirs d’application du gouvernement fédéral aux cas de spécification incorrecte des ingrédients du médicament.
En 1937, plus de 100 personnes sont mortes après avoir ingéré « Elixir Sulfanilamide » fabriqué par S.e. Société Massengill du Tennessee. Le produit a été formulé dans du diéthylène glycol, un solvant hautement toxique qui est maintenant largement utilisé comme antigel. En vertu des lois en vigueur à cette époque, les poursuites contre le fabricant n’étaient possibles que sous la technicité que le produit avait été appelé un « élixir », ce qui impliquait littéralement une solution dans l’éthanol. En réponse à cet épisode, le Congrès américain a adopté le Federal Food, Drug, and Cosmetic Act de 1938, qui exigeait pour la première fois une démonstration de sécurité avant la commercialisation d’un médicament et interdisait explicitement les fausses allégations thérapeutiques.
Les années d’après-guerre, 1945– 1970modifier
De nouveaux progrès dans la recherche anti-infectieuxmodifier
Au lendemain de la Seconde Guerre mondiale, la découverte de nouvelles classes de médicaments antibactériens, notamment les céphalosporines (développées par Eli Lilly sur la base des travaux fondateurs de Giuseppe Brotzu et Edward Abraham), la streptomycine (découverte lors d’un programme de recherche financé par Merck en Le laboratoire de Selman Waksman), les tétracyclines (découvertes aux laboratoires Lederle, qui font maintenant partie de Pfizer), l’érythromycine (découverte chez Eli Lilly and Co.) et leur extension à une gamme de plus en plus large d’agents pathogènes bactériens. La streptomycine, découverte lors d’un programme de recherche financé par Merck dans le laboratoire de Selman Waksman à Rutgers en 1943, est devenue le premier traitement efficace de la tuberculose. Au moment de sa découverte, les sanatoriums pour l’isolement des personnes infectées par la tuberculose étaient une caractéristique omniprésente des villes des pays développés, 50% mourant dans les 5 ans suivant leur admission.
Un rapport de la Federal Trade Commission publié en 1958 a tenté de quantifier l’effet du développement d’antibiotiques sur la santé publique américaine. Le rapport a constaté qu’au cours de la période 1946-1955, il y avait une baisse de 42% de l’incidence des maladies pour lesquelles les antibiotiques étaient efficaces et seulement une baisse de 20% de celles pour lesquelles les antibiotiques n’étaient pas efficaces. Le rapport conclut qu' »il semble que l’utilisation d’antibiotiques, le diagnostic précoce et d’autres facteurs aient limité la propagation de l’épidémie et donc le nombre de ces maladies qui se sont produites ». L’étude a examiné plus en détail les taux de mortalité de huit maladies courantes pour lesquelles des antibiotiques offraient un traitement efficace (syphilis, tuberculose, dysenterie, scarlatine, coqueluche, infections à méningocoques et pneumonie) et a constaté une baisse de 56% au cours de la même période. Parmi ceux-ci, on note une baisse de 75 % des décès dus à la tuberculose.
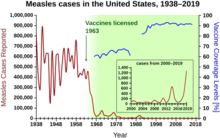
Au cours des années 1940-1955, le taux de baisse du taux de mortalité aux États-Unis s’est accéléré, passant de 2% par an à 8% par an, puis est revenu au taux historique de 2% par an. Le déclin spectaculaire de l’immédiat après-guerre a été attribué au développement rapide de nouveaux traitements et vaccins contre les maladies infectieuses qui s’est produit au cours de ces années.Le développement du vaccin a continué de s’accélérer, la réalisation la plus notable de la période étant le développement du vaccin contre la poliomyélite par Jonas Salk en 1954, grâce au financement de la Fondation nationale pour la paralysie infantile à but non lucratif. Le procédé vaccinal n’a jamais été breveté, mais a plutôt été donné à des sociétés pharmaceutiques pour qu’elles le fabriquent en tant que générique à faible coût. En 1960, Maurice Hilleman de Merck Sharp & Dohme a identifié le virus SV40, qui a ensuite été montré pour provoquer des tumeurs chez de nombreuses espèces de mammifères. Il a ensuite été déterminé que le SV40 était présent comme contaminant dans les lots de vaccins contre la poliomyélite administrés à 90 % des enfants aux États-Unis. La contamination semble provenir à la fois du stock de cellules d’origine et du tissu de singe utilisé pour la production. En 2004, l’Institut du cancer des États-Unis a annoncé qu’il avait conclu que le SV40 n’était pas associé au cancer chez les personnes.
D’autres nouveaux vaccins notables de la période comprennent ceux contre la rougeole (1962, John Franklin Enders du Children’s Medical Center Boston, plus tard affinés par Maurice Hilleman chez Merck), la rubéole (1969, Hilleman, Merck) et les oreillons (1967, Hilleman, Merck). Les incidences de la rubéole, du syndrome de rubéole congénitale, de la rougeole et des oreillons aux États-Unis ont toutes diminué de > 95% immédiatement après la vaccination généralisée. Les 20 premières années de vaccination autorisée contre la rougeole aux États-Unis ont permis d’éviter environ 52 millions de cas de la maladie, 17 400 cas de retard mental et 5 200 décès.
Développement et commercialisation de médicaments antihypertenseurs
L’hypertension est un facteur de risque d’athérosclérose, d’insuffisance cardiaque, de maladie coronarienne, d’ACCIDENT vasculaire cérébral, de maladie rénale et de maladie artérielle périphérique, et constitue le facteur de risque le plus important de morbidité et de mortalité cardiovasculaires, dans les pays industrialisés. Avant 1940, environ 23 % de tous les décès chez les personnes de plus de 50 ans étaient attribués à l’hypertension. Les cas graves d’hypertension ont été traités par chirurgie.
Les premiers développements dans le domaine du traitement de l’hypertension comprenaient des agents de blocage du système nerveux sympathique à l’ion ammonium quaternaire, mais ces composés n’ont jamais été largement utilisés en raison de leurs effets secondaires graves, car les conséquences à long terme de l’hypertension artérielle n’avaient pas encore été établies et parce qu’ils devaient être administrés par injection.
En 1952, des chercheurs de Ciba ont découvert le premier vasodilatateur disponible par voie orale, l’hydralazine. Une lacune majeure de l’hydralazine en monothérapie était qu’elle perdait son efficacité avec le temps (tachyphylaxie). Au milieu des années 1950, Karl H. Beyer, James M. Sprague, John E. Baer et Frederick C. Novello de Merck and Co. découvert et développé le chlorothiazide, qui reste le médicament antihypertenseur le plus utilisé aujourd’hui. Cette évolution a été associée à une baisse substantielle du taux de mortalité chez les personnes souffrant d’hypertension. Les inventeurs ont été reconnus par un Prix Lasker de la santé publique en 1975 pour « avoir sauvé des milliers de vies et allégé les souffrances de millions de victimes de l’hypertension ».
Une revue Cochrane de 2009 a conclu que les antihypertenseurs thiazidiques réduisent le risque de décès (RR 0,89), d’accident vasculaire cérébral (RR 0,63), de maladie coronarienne (RR 0,84) et d’événements cardiovasculaires (RR 0,70) chez les personnes souffrant d’hypertension artérielle. Au cours des années suivantes, d’autres classes d’antihypertenseurs ont été développées et ont été largement acceptées en association, notamment les diurétiques de l’anse (Lasix / furosémide, Hoechst Pharmaceuticals, 1963), les bêta-bloquants (ICI Pharmaceuticals, 1964) les inhibiteurs de l’ECA et les inhibiteurs des récepteurs de l’angiotensine. Les inhibiteurs de l’ECA réduisent le risque de nouvelle maladie rénale et de décès chez les patients diabétiques, qu’ils souffrent ou non d’hypertension.
Contraceptifs orauxmodifier
Avant la seconde guerre mondiale, le contrôle des naissances était interdit dans de nombreux pays et, aux États-Unis, même la discussion sur les méthodes contraceptives conduisait parfois à des poursuites en vertu des lois Comstock. L’histoire du développement des contraceptifs oraux est donc étroitement liée au mouvement de contrôle des naissances et aux efforts des militantes Margaret Sanger, Mary Dennett et Emma Goldman. Basé sur des recherches fondamentales effectuées par Gregory Pincus et des méthodes de synthèse de la progestérone développées par Carl Djerassi à Syntex et par Frank Colton à G.D. Searle & Co., le premier contraceptif oral, Enovid, a été développé par E.D. Searle and Co. et approuvé par la FDA en 1960. La formulation originale incorporait des doses excessives d’hormones et provoquait de graves effets secondaires. Néanmoins, en 1962, 1,2 million de femmes américaines prenaient la pilule et, en 1965, ce nombre était passé à 6,5 millions. La disponibilité d’une forme pratique de contraception temporaire a conduit à des changements radicaux dans les mœurs sociales, notamment en élargissant l’éventail des options de style de vie offertes aux femmes, en réduisant la dépendance des femmes à l’égard des hommes pour la pratique de la contraception, en encourageant le retard du mariage et en augmentant la cohabitation avant le mariage.
Thalidomide et les amendements Kefauver-Harris.

Aux États-Unis, une poussée pour des révisions de la Loi FD & C a émergé des audiences du Congrès dirigées par le sénateur Estes Kefauver du Tennessee en 1959. Les audiences ont porté sur un large éventail de questions de politique, y compris les abus de publicité, l’efficacité douteuse des médicaments et la nécessité d’une réglementation accrue de l’industrie. Alors que l’élan pour une nouvelle législation a temporairement été marqué par un débat prolongé, une nouvelle tragédie est apparue qui a souligné la nécessité d’une réglementation plus complète et a fourni la force motrice pour l’adoption de nouvelles lois.
Le 12 septembre 1960, un licencié américain, le William S. La société Merrell de Cincinnati a présenté une nouvelle demande de médicament pour le Kevadon (thalidomide), un sédatif commercialisé en Europe depuis 1956. Le médecin de la FDA en charge de l’examen du composé, Frances Kelsey, a estimé que les données à l’appui de l’innocuité de la thalidomide étaient incomplètes. L’entreprise a continué à faire pression sur Kelsey et la FDA pour qu’elle approuve la demande jusqu’en novembre 1961, date à laquelle le médicament a été retiré du marché allemand en raison de son association avec de graves anomalies congénitales. Plusieurs milliers de nouveau-nés en Europe et ailleurs ont subi les effets tératogènes de la thalidomide. Sans l’approbation de la FDA, l’entreprise a distribué du Kevadon à plus de 1 000 médecins sous couvert d’utilisation expérimentale. Plus de 20 000 Américains ont reçu de la thalidomide dans cette « étude », dont 624 patientes enceintes, et environ 17 nouveau-nés connus ont subi les effets du médicament.
La tragédie de la thalidomide a ressuscité le projet de loi de Kefauver visant à améliorer la réglementation des drogues qui avait achoppé au Congrès, et l’amendement Kefauver-Harris est devenu loi le 10 octobre 1962. Les fabricants devaient désormais prouver à la FDA que leurs médicaments étaient efficaces et sûrs avant de pouvoir être commercialisés sur le marché américain. La FDA a reçu le pouvoir de réglementer la publicité des médicaments sur ordonnance et d’établir de bonnes pratiques de fabrication. La loi exigeait que toutes les drogues introduites entre 1938 et 1962 soient efficaces. Une étude collaborative FDA-Académie nationale des Sciences a montré que près de 40% de ces produits n’étaient pas efficaces. Une étude aussi approfondie des produits en vente libre a commencé dix ans plus tard.
1970–1980sEdit
Statinesedit
En 1971, Akira Endo, un biochimiste japonais travaillant pour la société pharmaceutique Sankyo, a identifié la mévastatine (ML-236B), une molécule produite par le champignon Penicillium citrinum, comme un inhibiteur de l’HMG-COA réductase, une enzyme critique utilisée par l’organisme pour produire du cholestérol. Les essais chez l’animal ont montré un très bon effet inhibiteur comme dans les essais cliniques, mais une étude à long terme chez le chien a révélé des effets toxiques à des doses plus élevées et, par conséquent, la mévastatine était considérée comme trop toxique pour un usage humain. La mévastatine n’a jamais été commercialisée, en raison de ses effets indésirables de tumeurs, de détérioration musculaire et parfois de mort chez les chiens de laboratoire.
P. Roy Vagelos, scientifique en chef et plus tard PDG de Merck & Co, s’est intéressé et a effectué plusieurs voyages au Japon à partir de 1975. En 1978, Merck avait isolé la lovastatine (mevinolin, MK803) du champignon Aspergillus terreus, commercialisé pour la première fois en 1987 sous le nom de Mevacor.
En avril 1994, les résultats d’une étude parrainée par Merck, la Scandinavian Simvastatin Survival Study, ont été annoncés. Les chercheurs ont testé la simvastatine, vendue plus tard par Merck sous le nom de Zocor, sur 4 444 patients présentant un taux de cholestérol élevé et une maladie cardiaque. Après cinq ans, l’étude a conclu que les patients avaient vu une réduction de 35% de leur cholestérol et que leurs chances de mourir d’une crise cardiaque étaient réduites de 42%. En 1995, Zocor et Mevacor ont tous deux fait gagner plus de 1 milliard de dollars à Merck. Endo a reçu le Prix du Japon en 2006 et le Prix de recherche Médicale clinique Lasker-DeBakey en 2008. Pour ses « recherches pionnières sur une nouvelle classe de molécules » pour « abaisser le cholestérol « ,