Résumé
Les tumeurs surrénales peuvent être bénignes ou impliquer un niveau élevé de morbidité et de mortalité dû à leur activité hormonale, ou à une éventuelle histologie maligne. La littérature médicale indique que cette condition est due à l’amélioration de la technique et à la diffusion dans un large éventail de méthodes d’imagerie, augmentant considérablement le diagnostic des nodules surrénaliens et son traitement immédiat. Lors du traitement d’un patient atteint de tumeurs surrénales, la principale préoccupation du professionnel doit être d’établir si la lésion est un néoplasme malin et s’il existe un fonctionnement hormonal, qui sont deux occurrences nécessitant généralement une intervention chirurgicale. Pour déterminer si une lésion fonctionne hormonalement, il est nécessaire d’effectuer des évaluations cliniques et endocriniennes complètes. Ainsi, il devrait faire partie de la routine du médecin généraliste de reconnaître et d’évaluer si la lésion est maligne ou fonctionnelle, situations dans lesquelles une surrénalectomie est nécessaire.
Keywords
Adrenocortical tumors, Adenoma, Adrenal, Cushing’s syndrome, Aldosteronoma
List of Acronyms
ACTH: Adrenocorticotropic Hormone; DEXA: Dexamethasone; FEO: Pheochromocytoma; FEO/ PGL: Feocromocitoma/Paraganglioma; HA: Systemic Arterial Hypertension; SAGH: Autonomous Subclinical Hypersecretion of Glucocorticoids; SC: Serum Cortisol; UFC: Urine Free Cortisol; IA: Adrenal Incidentaloma; HU: Hounsfield Unit
Introduction
Adrenal gland tumors are common diseases in clinical practices. Selon la façon dont ils se manifestent, ils sont classés en fonctionnants (qui produisent des hormones) et non fonctionnels (également appelés silencieux). En termes de comportement biologique, ils peuvent être divisés en tumeurs bénignes ou malignes.
La plupart des tumeurs corticosurrénales sont des adénomes bénins, unilatéraux et non fonctionnels, présentant un diamètre inférieur à 4 cm, perçus lors d’études d’imagerie abdominale.
Cette « découverte fortuite » porte le nom d’accidentalome surrénalien et est définie comme une masse surrénale, cliniquement insoupçonnée, qui se retrouve dans des études d’imagerie menées pour d’autres raisons que l’évaluation des glandes surrénales. Ces dernières années, la prévalence des adénomes surrénaliens a augmenté en raison de l’utilisation de l’imagerie abdominale avec une sensibilité croissante.
Les tumeurs surrénaliennes fonctionnelles sont généralement de type adénome bénin, ce qui provoque, par exemple, le syndrome de Cushing, l’aldostéronisme primaire ou, plus rarement, la virilisation. Dans la moelle osseuse, les phéochromocytomes, tumeurs sécrétoires des catécholamines, qui se distinguent bien qu’elles soient rares et impliquent une grande morbidité et mortalité.
Épidémiologie
Dans une étude combinée avec des critères de sélection et de diagnostic différents, nous avons eu les principales étiologies des tumeurs surrénales: Adénomes 41%, Métastases 19%, Carcinomes 10%, Myélolipomes 9% et Phéochromocytomes 8%. La plupart des cas restants (13%) étaient des lésions bénignes, telles que des kystes surrénaliens.
Dans les rapports cliniques, les cas d’incidence surrénalienne présentent un pic d’incidence entre la cinquième et la septième décennie. L’âge moyen des patients au moment du diagnostic est de 55 ans, sans différence significative d’âge entre les femmes et les hommes.
La prévalence augmente avec l’âge; le taux est inférieur à 1% chez les patients de moins de 30 ans et à 7% chez les patients de 70 ans et plus.
Attribué à la production hormonale, bien que la plupart des tumeurs ne fonctionnent pas, une production légèrement accrue de certaines hormones peut être vérifiée dans au moins 15% des cas.
Évaluation hormonale
Les adénomes ou carcinomes surrénaliens sécrétant du cortisol, les phéochromocytomes, les aldostéronomes et les lésions sécrétoires des androgènes sont les types de masses surrénales sécrétoires ou fonctionnelles diagnostiquées de manière plus récurrente.
Tumeurs productrices de cortisol
Ces tumeurs produisent généralement des quantités réduites de cortisol, qui, dans la plupart des cas, ne sont pas suffisantes pour augmenter l’excrétion de cortisol libre dans l’urine. Ils sont cependant capables de provoquer la suppression de l’axe hypothalamo-hypophysaire. Habituellement, il n’y a pas de manifestations liées au syndrome de Cushing chez les patients. Pour cette raison, cette condition est connue sous le nom de syndrome de Cushing subclinique ou hypercortisolisme subclinique.
Il est important de souligner la différence entre le syndrome de Cushing subclinique, caractérisé par une anomalie biochimique cliniquement non manifestée et le syndrome de Cushing préclinique, qui est le stade initial du développement du syndrome lui-même. L’hypersécrétion subclinique autonome des glucocorticoïdes (SAGH) est le terme le plus courant, proposé pour définir une sécrétion autonome de cortisol par un adénome surrénalien chez les patients sans symptômes du syndrome de Cushing.
Chez l’adulte, les signes et symptômes les plus suggestifs de la présence d’hypercortisolisme comprennent une faiblesse musculaire proximale, une pléthore faciale, une perte de membres avec une augmentation de la graisse dans l’abdomen et le visage, de larges stries pourpres, des hématomes sans traumatisme évident et des compresses supraclaviculaires.
Cependant, en raison de nombreux symptômes d’hypercortisolisme, parmi eux, l’hypertension et le diabète ne sont pas caractéristiques, et le degré de leur apparence clinique est compatible avec la variation de l’étendue de la surproduction hormonale, l’indication précise de la prévalence de la SAGH, sera soumise aux résultats obtenus par les méthodes des tests utilisés et aux critères de sélection des patients symptomatiques pour la confirmation de la maladie.
Lorsque vous suspectez le syndrome de Cushing, un test de suppression de la dexaméthasone à 1 mg doit être effectué pendant la nuit. La veille de la collecte du cortisol sérique à 8 heures, le patient prend 1 mg de dexaméthasone par voie orale à 23 heures.
Il est considéré comme une réponse anormale au cortisol lorsqu’elle est présentée dans la gamme de 1,8 à 5,0 mcg / dl, ou plus précisément:
A) Si
B) Si entre 1,8 et 5 mcg / dL: Résultats indéterminés;
C) > 5 mcg / dL: Diagnostic hautement probable du syndrome de Cushing;
Une suppression anormale avec de la dexaméthasone équivaut à un dépistage suspect, elle doit donc être confirmée par une dose de cortisol urinaire libre de 24 heures, après administration de 8 mg de dexaméthasone pendant la nuit et une dose sérique d’ACTH, afin de pouvoir déterminer l’origine du syndrome de Cushing, qui apparaît généralement avec des niveaux non comprimés (dépendants de l’ACTH).
Dans le cas du diagnostic du syndrome de Cushing subclinique, les critères suivants sont utilisés:
– Cortisol plasmatique > 5 mcg / dL dans le test de dexaméthasone à 1 mg sans autre stigmate ou au moins 2 des résultats suivants.
1.ACTH plasmatique
2.Le cortisol urinaire libre dans un échantillon de 24 heures a augmenté;
3.Cortisol sérique > 3 mcg / dL dans le test de dexaméthasone à 1 mg (figure 1).
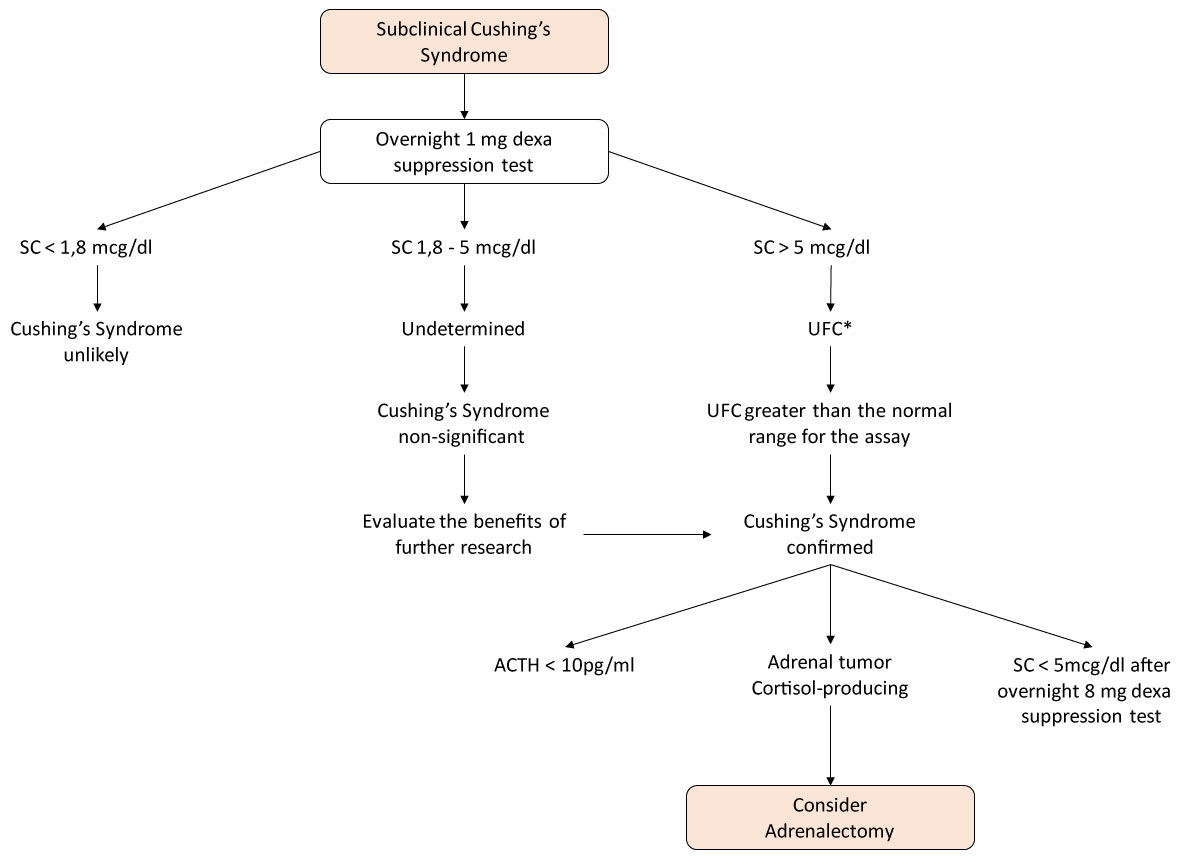 Figure 1: Algorithme d’investigation du syndrome de Cushing dans les cas de Surrénales. * Au moins deux mesures. Voir La Figure 1
Figure 1: Algorithme d’investigation du syndrome de Cushing dans les cas de Surrénales. * Au moins deux mesures. Voir La Figure 1
Ainsi, il est difficile de parvenir à un consensus pour l’approche du syndrome de Cushing subclinique, qui peut être traité cliniquement ou par chirurgie. Pour les patients présentant de nombreuses comorbidités pouvant être liées à l’hypercortisolisme, le rapport bénéfice / risque de l’surrénalectomie doit être considéré comme un traitement. Une grande partie des patients subissant cette chirurgie peut développer une insuffisance surrénale aiguë (parfois fatale), il est donc extrêmement important de couvrir périopératoire avec administration de glucocorticoïdes.
Tumeurs productrices de catécholamines
Les phéochromocytomes sont des tumeurs de cellules chromaffines de la médullosurrénale qui produisent, stockent, métabolisent et sécrètent des catécholamines. dans certains cas, les paragangliomes d’autres hormones peptidiques (PGL) sont des tumeurs similaires, mais d’origine supplémentaire. Le syndrome Feo / PGL est une maladie rare, avec une prévalence estimée entre 0,1 et 0,2% de la population de personnes hypertendues.
La plupart des tumeurs sécrétant des catécholamines sont sporadiques. Cependant, certains patients (environ 40%) ont la maladie dans le cadre d’un trouble familial; chez ces patients, les tumeurs sécrétant des catécholamines sont plus susceptibles d’être des phéochromocytomes surrénaliens bilatéraux ou des paragangliomes.
Il existe plusieurs troubles familiaux associés au phéochromocytome, tous à transmission autosomique dominante:
• Syndrome de Von Hippel-Lindau (VHL), associé à des mutations du gène suppresseur de tumeur VHL.
• Néoplasme multiple endocrinien de type 2 (MEN2), associé à des mutations du proto-oncogène RET.
• Un phéochromocytome est également observé, bien que rarement, de neurofibromatose de type 1 (NF1), due à des mutations du gène NF1.
Ces tumeurs revêtent une importance particulière car, bien que rares (ainsi que les adénomes sécrétoires de l’aldostérone) donnent lieu à une forme d’hypertension corrigible chirurgicalement. L’hypertension est souvent paroxystique. La triade classique des symptômes chez les patients atteints de phéochromocytome se compose de maux de tête épisodiques, de transpiration et de tachycardie, et il existe une prévalence de patients qui ne présentent pas les trois symptômes classiques. Outre les patients présentant les manifestations cliniques typiques de la maladie, l’investigation doit également être effectuée chez des personnes ayant des antécédents familiaux de FEO ou de carcinome médullaire de la thyroïde, en présence d’HA chez les jeunes, d’HA de contrôle difficile ou d’induction anesthésique.
Le diagnostic de phéochromocytome est généralement posé par des mesures de métanéphrines et de catécholamines fracturées dans l’urine et le plasma, comme le montre la figure 2.
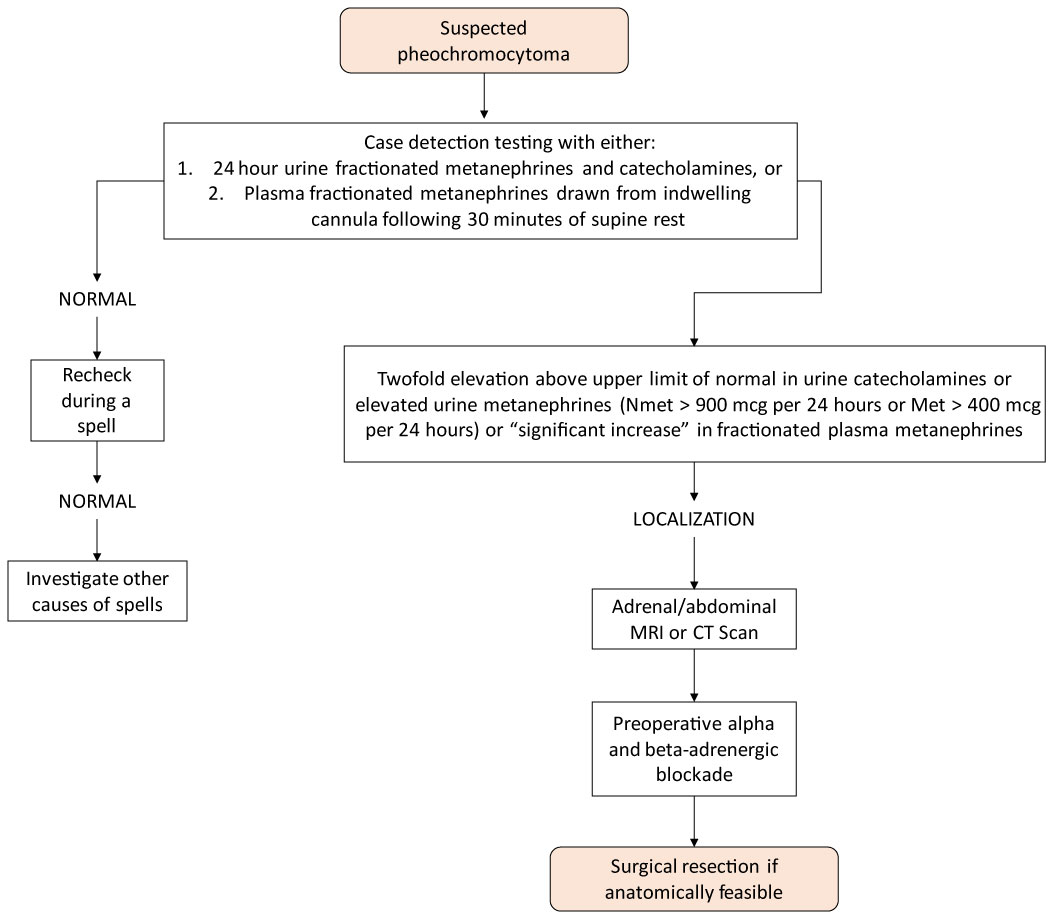 Figure 2 : Algorithme d’investigation des phéochromocytomes dans les cas de Surrénales. Voir La Figure 2
Figure 2 : Algorithme d’investigation des phéochromocytomes dans les cas de Surrénales. Voir La Figure 2
En ce qui concerne les méthodes d’image, les phéochromocytomes ont une plus grande atténuation sur la tomodensitométrie non améliorée (> 20 HU), une vascularisation accrue de la masse, un retard dans le lavage du produit de contraste (10 minutes après l’administration du produit de contraste, un lavage absolu du produit de contraste inférieur à 50%), une force de signal élevée sur l’IRM pondérée T2, des changements kystiques et hémorragiques et une taille variable et peuvent être bilatéraux.
En revanche, les caractéristiques d’imagerie qui suggèrent un carcinome surrénalien ou des métastases comprennent: Forme irrégulière, densité inhomogène, valeurs élevées d’atténuation CT non améliorées (> 20 HU), délavage retardé du produit de contraste (par exemple, 4 cm et calcification tumorale.
Tumeurs productrices d’aldostérone
Les aldostéronomes sont rares et difficiles à détecter et se caractérisent cliniquement par une hypertension artérielle systémique liée à une hypokaliémie. Étant donné que la plupart des patients sont asymptomatiques en ce qui concerne l’hypocalcémie, c’est-à-dire qu’ils sont normocaliémiques, il est recommandé d’évaluer tous les patients souffrant d’hypertension associée à une incidence surrénalienne en mesurant leur concentration plasmatique d’activité plasmatique de l’aldostérone et de la rénine et le rapport entre eux supérieur à 30 suggère fortement une production autonome d’aldostérone. Une raison plus grande que 50 distingue clairement l’aldostéronisme primaire des autres formes d’hypertension essentielle déjà un résultat inférieur à 20 conteste le diagnostic et les résultats dans l’intervalle entre 20 et 30, indiquent un diagnostic plus précis.
Les patients qui utilisent la spironolactone ne peuvent pas être évalués par la relation aldostérone et l’activité plasmatique. D’autres médicaments qui peuvent contribuer à des résultats douteux sont les bêta-bloquants, l’agoniste alpha-adrénergique central et les anti-inflammatoires. De même, certains médicaments peuvent présenter une réduction de l’inhibiteur de l’enzyme conservatrice de l’angiotensine, tels que le bloqueur des récepteurs de l’aldostérone, les inhibiteurs de la thiazine et la dihydropyridine du canal calcique Figure 3.
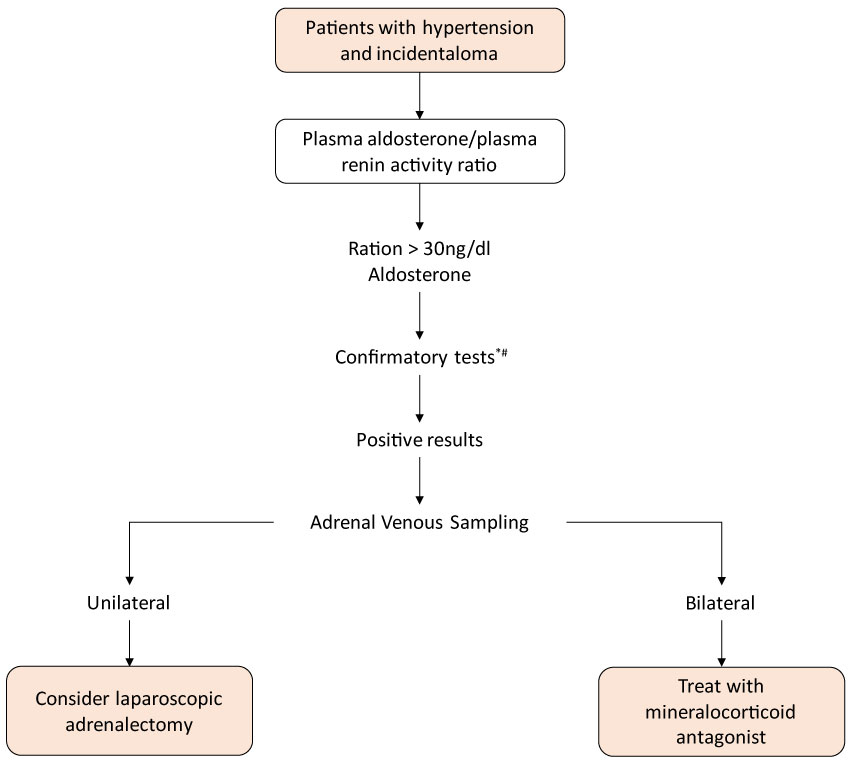 Figure 3: Algorithme d’investigation de l’hyperaldostéronisme dans les cas de Surrénales. * Tests de confirmation les plus couramment utilisés: Test de charge en sodium par voie orale, test de perfusion intraveineuse de solution saline. Voir La Figure 3
Figure 3: Algorithme d’investigation de l’hyperaldostéronisme dans les cas de Surrénales. * Tests de confirmation les plus couramment utilisés: Test de charge en sodium par voie orale, test de perfusion intraveineuse de solution saline. Voir La Figure 3
Le cathétérisme surrénalien est la méthode d’évaluation permettant de vérifier si l’augmentation de la production d’aldostérone chez les patients de plus de 40 ans, avec hyperaldostéronisme confirmé, est réellement causée par un accidentalome ou une hyperplasie surrénalienne.
Dans de tels cas, l’surrénalectomie ne résoudrait pas l’hyperproduction hormonale, qui devrait être contrôlée avec l’utilisation de médicaments, des antagonistes de l’aldostérone tels que la spironolactone.
Chez les patients présentant une hypokaliémie spontanée, une rénine plasmatique inférieure aux niveaux de détection et une aldostérone plasmatique > 20 ng / dl, il est suggéré qu’il n’y a pas besoin de tests de confirmation supplémentaires.
Tumeurs produisant des androgènes et des œstrogènes
En cas d’hyperplasie surrénalienne congénitale due à un déficit en 21-hydroxylase, il est courant de trouver des masses surrénales, uni ou bilatérales, à la suite de la stimulation excessive chronique des surrénales par l’ACTH.
L’hormone sexuelle qui produit des adénomes surrénaliens est très rare. Les carcinomes producteurs d’androgènes sont également rares. Les cas d’androgènes ou d’excès d’œstrogènes sont rarement décrits chez les patients présentant des adénomes corticosurrénaux bénins, mais, en général, ils se manifestent par des symptômes ou des signes de virilisation chez la femme (acné, hirsutisme) ou de féminisation chez l’homme (Gynécomastie). Ainsi, de telles lésions ne peuvent pas être considérées comme de véritables IAs. Par conséquent, la nécessité de mesurer les hormones sexuelles et les précurseurs de stéroïdes est limitée en cas de lésions surrénales avec indétermination ou suspicion de caractéristiques d’imagerie de la malignité, où des niveaux élevés peuvent indiquer l’origine corticosurrénale de la tumeur et suggérer la présence d’un adénocarcinome surrénalien. L’surrénalectomie est indiquée pour le contrôle des hormones chez les individus présentant une virilisation ou des concentrations élevées d’androgènes.
Les tumeurs productrices d’œstrogènes sont rares et, dans la plupart des cas, malignes. La présence de ces tumeurs chez les hommes se manifeste généralement par une féminisation avec gynécomastie, une diminution de la libido, une atrophie des testicules; chez les femmes, elle peut se manifester par une sensibilité mammaire et des saignements. Dans de tels cas, une surrénalectomie peut également être indiquée.
Discussion
Les études d’investigation de l’imagerie surrénale sont particulièrement pertinentes pour comprendre que le professionnel de ce domaine est attentif et curieux de tout trait d’anomalie suspectant la santé des glandes surrénales.
En parallèle, on peut observer l’importance de la connaissance des méthodes efficaces de diagnostic et de traitement pertinentes à chaque cas détecté.
De tous les protocoles utilisés et proposés pour l’investigation hormonale, celui avec la récidive la plus élevée est celui qui utilise du cortisol sérique après suspension nocturne, avec 1 mg de dexaméthasone (pour l’hypercortisolisme subclinique) et des métanéphrines plasmatiques (pour la FEO). L’investigation des aldostéronomes n’est indiquée que pour les cas d’hypertension et / ou d’hypokaliémie, la détermination de l’activité plasmatique de l’aldostérone et de la rénine plasmatique.
De nombreuses études publiées et diffusées dans des colloques médicaux mentionnent plusieurs protocoles utilisés pour l’investigation hormonale, l’observation et la découverte d’anomalies des cellules tissulaires des glandes surrénales, les différentes tumeurs que ces Glandes peuvent abriter et des mesures préventives et thérapeutiques des maladies, offrant une qualité de vie aux patients.
Remerciements / Conflits d’intérêts
Aucun des auteurs n’a de conflit d’intérêts.
Citation
do Prado BC, Schafascheck GS, Puppim AR (2019) Évaluation hormonale des tumeurs surrénales: Ce que le Médecin généraliste Doit Savoir. Int Arch Urol Complic 5:063. doi.org/10.23937/2469-5742/1510063