MAE. Ci sono molte distinte sindromi di epilessia infantile di cui siamo diventati criticamente consapevoli nell’era genomica in quanto sono collegate a cause genetiche prominenti, tra cui la sindrome di Dravet (SCN1A) e l’epilessia dell’infanzia con crisi focali migratorie (KCNT1). Tuttavia, ci sono molte altre sindromi epilettiche in cui una causa genetica è stata a lungo sospettata, ma è rimasta inafferrabile. Una delle sindromi epilettiche che è rimasta in gran parte inesplorata è la sindrome di Doose, indicata anche come epilessia mioclonica astatica (MAE) o epilessia con convulsioni miocloniche-atoniche. In un recente studio sull’epilessia, abbiamo esplorato l’architettura genetica della sindrome di Doose e identificato le cause monogeniche nel 14% degli individui, tra cui SYNGAP1, NEXMIF (KIAA2022) e SLC6A1. Il nostro studio suggerisce che la sindrome di Doose è geneticamente eterogenea, possibilmente con un paesaggio genetico distinto.
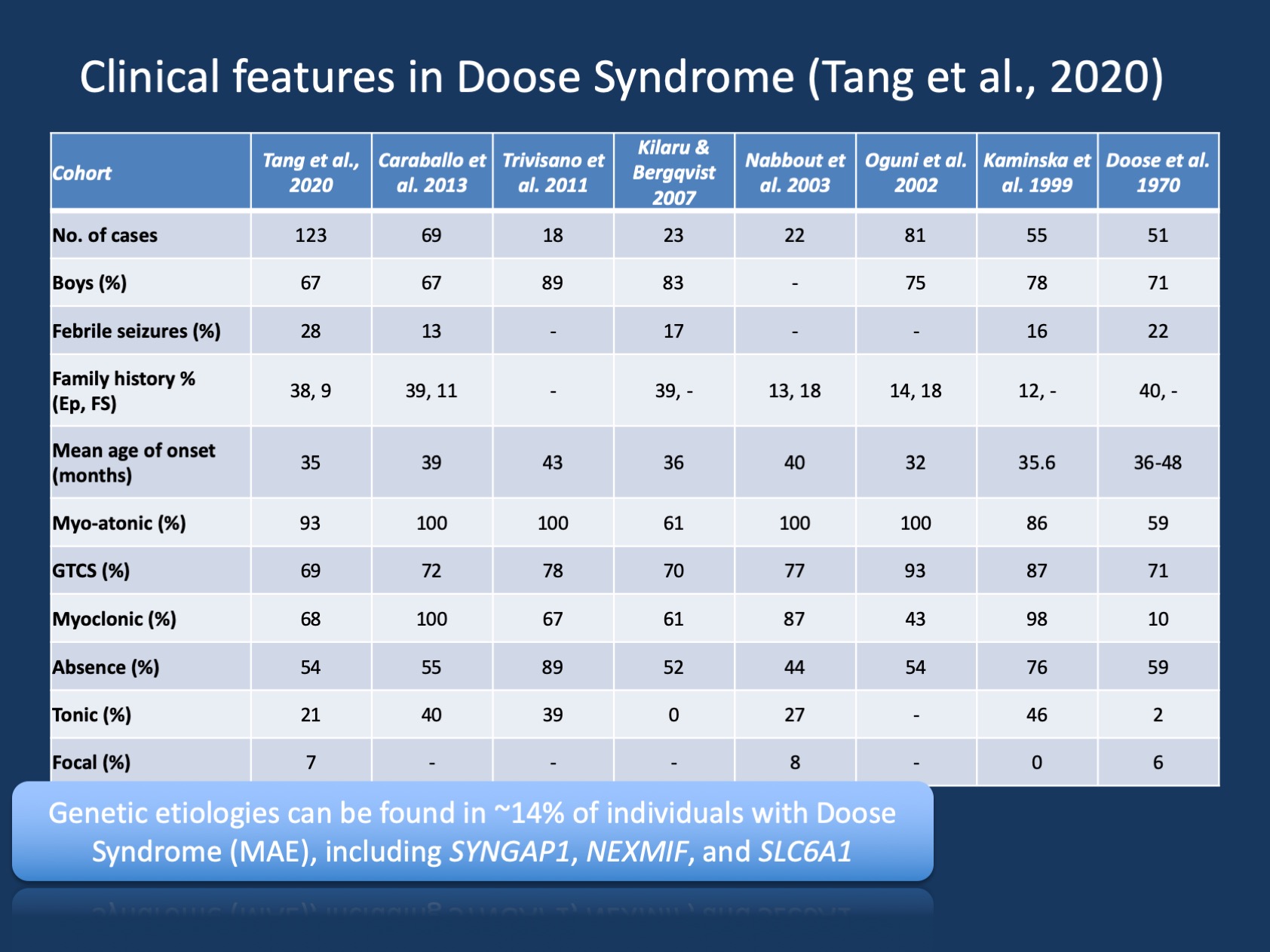
Figura 1. Le caratteristiche cliniche della sindrome di Doose (epilessia con convulsioni atoniche miocloniche o epilessia astatica mioclonica ). La tabella è adattata dalla nostra recente pubblicazione di Tang et al., 2020 e confronta le caratteristiche cliniche nelle grandi coorti della sindrome di Doose che sono state riferite dalla descrizione iniziale nel 1970. La sindrome di Doose (MAE) è una sindrome da epilessia rara, ma è un’entità clinica riconoscibile. Tuttavia, in contrasto con la sindrome di Dravet dove >90% degli individui hanno varianti che causano malattie in SCN1A, la sindrome di Doose è geneticamente eterogenea.
La storia di MAE. Prima di tutto, mi permetta di chiarire alcuni problemi di denominazione. La sindrome di Doose o epilessia mioclonica astatica (MAE) è ora ufficialmente indicata come Epilessia con crisi miocloniche-atoniche utilizzando la più recente convenzione di denominazione. Tuttavia, mentre in genere cerchiamo di allinearci con il modo ufficiale di come vengono utilizzati i termini legati all’epilessia, permettetemi di essere un po ‘ testardo qui. Come neurologo infantile addestrato presso il Dipartimento di Neuropediatria a Kiel, in Germania, mi riferirò a questa condizione come Sindrome di Doose usando MAE come stenografia, onorando Hermann Doose (1927-2018) che è uno dei fondatori dell’epilettologia pediatrica europea (link a ILAE in memoriam). Molti madrelingua inglesi in genere sbagliano la pronuncia – la pronuncia corretta è “Dohs-ah “e NON fa rima con”goose”.
Il quadro clinico di MAE. Doose è in genere accreditato di fare un’importante osservazione clinica: negli anni ‘ 60, lui e i suoi collaboratori a Kiel riconobbero un’epilessia insolita nei bambini caratterizzata da attacchi improvvisi di caduta (convulsioni atoniche), convulsioni miocloniche, convulsioni tonico-cloniche generalizzate e un pattern EEG generalizzato, che si verificano tipicamente tra i due ei cinque anni. Questa epilessia si è verificata in genere nei bambini con uno sviluppo tipico precedente (vedi sotto) e spesso ha portato a ritardo dello sviluppo e disabilità intellettiva. Doose e collaboratori hanno riconosciuto che questa sindrome da epilessia recentemente riconosciuta era diversa dalla sindrome di Lennox-Gastaut o epilessie con convulsioni tonico-cloniche generalizzate precipitate dalla febbre, le ultime delle quali sono state successivamente concettualizzate come sindrome di Dravet.
Capire MAE. Le descrizioni cliniche della sindrome di Doose (MAE) sono state costanti nel corso degli anni, ma MAE non ha ricevuto tanta attenzione quanto le sindromi da epilessia infantile in cui è stata scoperta una base genetica e in cui la ricerca mirata può consentire nuovi trattamenti. Nella nostra recente pubblicazione di Tang e collaboratori, abbiamo elencato otto studi negli ultimi 50 anni tra il 1970 e il 2020 con un totale di individui descritti 442. Di conseguenza, MAE è raro, ma è abbastanza comune che la maggior parte dei neurologi infantili e degli epilettologi pediatrici siano consapevoli di questa condizione. Solo un piccolo sottoinsieme di individui con MAE ha risultati cerebrali strutturali o anomalie metaboliche che spiegano l’epilessia sottostante – nella stragrande maggioranza dei bambini con MAE, neuroimaging e metabolica work-up è insignificante.
Trovare la causa di MAE. L’assenza di imaging esplicativo o risultati metabolici solleva la preoccupazione per una causa genetica sottostante. Mentre studi precedenti avevano cercato di fissare un contributo genetico attraverso studi epidemiologici valutando le storie familiari, questo metodo tradizionale è stato in gran parte sostituito da recenti scoperte nella genetica dell’epilessia. Valutare la base genetica delle epilessie infantili gravi è ora fatto attraverso la genetica molecolare, piuttosto che epidemiologia genetica. Valutare l’architettura genetica di MAE è stato uno degli obiettivi del nostro studio di Tang e collaboratori pubblicato su Epilepsia.
MAE è una malattia? Prima di descrivere i risultati genetici in MAE, permettetemi di introdurre un dibattito in corso all’interno del campo MAE. La descrizione iniziale di Doose e collaboratori includeva in gran parte bambini con sviluppo tipico prima dell’inizio delle crisi (>75%), che è paragonabile al nostro recente studio (79%). Tuttavia, quando si confrontano i bambini con e senza precedenti problemi di sviluppo, stiamo guardando la stessa malattia o solo i bambini con un precedente sviluppo insignificante dovrebbero essere considerati tipici MAE? Mentre questa domanda non è facilmente risolta quando si considerano le storie e gli esiti dell’epilessia, le alterazioni genetiche sottostanti possono fornire alcuni indizi. Le cause genetiche sono identificate più frequentemente in bambini con i disordini neurodevelopmental più severi e tutti i bambini con le cause genetiche identificate hanno avuti disordini neurodevelopmental complessi oltre all’epilessia. Di conseguenza, le cause genetiche possono essere osservate nei bambini con MAE, ma in genere solo nei bambini con MAE e disturbi dello sviluppo neurologico comorbido, suggerendo che MAE non è solo una condizione geneticamente eterogenea, ma ha caratteristiche simili ad altre epilessia nell’infanzia rispetto alla loro eterogeneità genetica e comorbidità. Ad esempio, nella sindrome di West (spasmi infantili), le cause genetiche sono anche identificate solo nei bambini con risultati dello sviluppo neurologico e non nei bambini che rispondono direttamente ai farmaci senza sequele dello sviluppo neurologico.
Scoperte genetiche in MAE. Lo spettro genetico in MAE è diverso rispetto alle encefalopatie epilettiche in generale. Il test genetico è stato eseguito in 85 individui e 12/85 (14%) individui hanno avuto risultati genetici positivi. Tre geni sono stati trovati in due individui, tra cui SYNGAP1, NEXMIF (KIAA2022) e SLC6A1. Questa triade di geni è stata precedentemente collegata a encefalopatie evolutive ed epilettiche con convulsioni generalizzate. Inoltre, singoli individui sono stati trovati con varianti che causano malattie in KCNA2, SCN2A, STX1B, KCNB1 e MECP2. Tutti questi geni sono stati precedentemente discussi sul nostro blog e sono noti per presentare con convulsioni generalizzate e caratteristiche EEG generalizzate.
Geni assenti in MAE. Mentre alcuni rapporti precedenti hanno trovato varianti in SCN1A, questo non è stato trovato nella nostra coorte. Inoltre, altri geni comuni di epilessia sono notevolmente assenti, tra cui CDKL5 e SLC2A1 (GLUT1). Ciò suggerisce che il panorama genetico di MAE è in qualche modo distinto e diverso da altre encefalopatie evolutive ed epilettiche, anche se solo conclusioni limitate possono essere tratte da coorti relativamente piccole. Il rendimento diagnostico in MAE non è molto alto-il 14% è molto più basso di quello tipicamente visto nelle encefalopatie evolutive ed epilettiche. Tuttavia, quando si escludono gli individui con sviluppo tipico, i rendimenti diagnostici sono molto più alti, suggerendo che il test genetico è di valore nei bambini con MAE. In generale, anche se nessun singolo individuo con MAE ha avuto varianti che causano la malattia in SLC2A1, test genetici per le cause trattabili di epilessie genetiche possono generalmente essere considerati in epilessie generalizzate atipiche.
L’ereditabilità mancante di MAE. Se il test genetico è negativo per MAE in più dell ‘ 80% degli individui, dov’è il carico genetico nascosto? A partire dal 2020, suggerirei due possibili spiegazioni. In primo luogo, il carico genetico delle epilessie generalizzate non è completamente sconosciuto. Più del 30% del rischio di popolazione per condizioni come l’epilessia dell’assenza infantile (CAE) o l’epilessia mioclonica giovanile (JME) è spiegato. Tuttavia, questa spiegazione non è data da cause a singolo gene, ma dal rischio poligenico, riferendosi all’effetto additivo di migliaia di varianti genetiche comuni. Si potrebbe ipotizzare che il MAE possa rappresentare un “fenotipo estremo” di epilessie generalizzate dove questo rischio poligenico è particolarmente elevato. Questa ipotesi è verificabile e sarà sicuramente perseguita in futuro. Inoltre, c’è di più nel genoma umano rispetto all’esoma – recenti scoperte nelle epilessie hanno sottolineato il ruolo delle varianti non codificanti, come le espansioni ripetute nell’epilessia mioclonica adulta familiare (FAME). Date le caratteristiche cliniche sorprendentemente simili in molti bambini con MAE, può ancora essere ragionevole cercare una causa genetica comune per molti bambini con MAE al di fuori dell’esoma, utilizzando il sequenziamento dell’intero genoma. Prendendo insieme, la valutazione dei punteggi di rischio poligenico (PRS) e il sequenziamento dell’intero genoma (WGS) sono due potenziali vie per identificare ulteriormente la base genetica del MAE.
Cosa devi sapere. La sindrome di Doose (MAE) è una sindrome da epilessia presumibilmente genetica con una causa eterogenea. Le varianti che causano malattie sono presenti nel 14% dei bambini e sono tipicamente identificate in bambini con ulteriori caratteristiche dello sviluppo neurologico come ritardo dello sviluppo o autismo. Una triade di geni tra cui SYNGAP1, NEXMIF (KIAA2022) e SLC6A1, sono cause genetiche ricorrenti che possono suggerire una biologia sottostante condivisa che è in qualche modo distinta da altre encefalopatie evolutive ed epilettiche.

Ingo Helbig è un neurologo infantile e ricercatore di genetica dell’epilessia che lavora presso l’Ospedale pediatrico di Philadelphia (CHOP), USA. Guida anche il gruppo di genetica dell’epilessia presso l’Università di Kiel, in Germania.

