Abstract
I tumori surrenali possono essere una scoperta benigna o implicare un alto livello di morbilità e mortalità a causa della loro attività ormonale o di una possibile istologia maligna. La letteratura medica indica che questa condizione è dovuta al miglioramento della tecnica e alla diffusione in una vasta gamma di metodi di imaging, aumentando considerevolmente la diagnosi dei noduli surrenali e il suo trattamento immediato. Quando si tratta di un paziente con tumori surrenali, la preoccupazione principale del professionista dovrebbe essere quella di stabilire se la lesione è una neoplasia maligna e se c’è qualche funzionamento ormonale, che sono due eventi che di solito richiedono un intervento chirurgico. Per stabilire se una lesione funziona ormonalmente è necessario eseguire valutazioni cliniche ed endocrine complete. Pertanto, dovrebbe essere parte della routine del medico generico riconoscere e valutare se la lesione è maligna o funzionante, situazioni in cui è necessaria l’adrenalectomia.
Keywords
Adrenocortical tumors, Adenoma, Adrenal, Cushing’s syndrome, Aldosteronoma
List of Acronyms
ACTH: Adrenocorticotropic Hormone; DEXA: Dexamethasone; FEO: Pheochromocytoma; FEO/ PGL: Feocromocitoma/Paraganglioma; HA: Systemic Arterial Hypertension; SAGH: Autonomous Subclinical Hypersecretion of Glucocorticoids; SC: Serum Cortisol; UFC: Urine Free Cortisol; IA: Adrenal Incidentaloma; HU: Hounsfield Unit
Introduction
Adrenal gland tumors are common diseases in clinical practices. Secondo il modo in cui si manifestano, sono classificati in funzionanti (che producono ormoni) e non funzionanti (noti anche come silenziatori). In termini di comportamento biologico, possono essere suddivisi in tumori benigni o maligni .
La maggior parte dei tumori adrenocorticali sono adenomi benigni, unilaterali e non funzionanti, che presentano meno di 4 cm di diametro, percepiti durante gli studi di imaging addominale .
Questo “reperto incidentale” è dato il nome di incidentaloma surrenale ed è definito come una massa surrenale, clinicamente insospettata, che si trova negli studi di imaging condotti per altri motivi rispetto alla valutazione delle ghiandole surrenali. Negli ultimi anni, la prevalenza di adenomi surrenali è aumentata a causa dell’uso di imaging addominale con crescente sensibilità .
I tumori surrenali funzionanti sono di solito del tipo di adenoma benigno, che causa, ad esempio, la sindrome di Cushing, l’aldosteronismo primario o, non così comunemente, la virilizzazione . Nel midollo osseo, feocromocitomi, tumori secretori delle catecolamine, che si distinguono pur essendo rari e implicano in grande morbilità e mortalità .
Epidemiologia
In uno studio combinato con diversi criteri di selezione e diagnosi, abbiamo avuto le principali eziologie dei tumori surrenali: Adenomi 41%, Metastasi 19%, Carcinomi 10%, Mielolipomi 9% e Feocromocitomi 8%. La maggior parte dei casi rimanenti (13%) erano lesioni benigne, come le cisti surrenali .
Nei rapporti clinici, gli incidentalomi surrenali mostrano un’incidenza di picco nella quinta-settima decade. L’età media dei pazienti alla diagnosi è di 55 anni, senza differenze significative di età tra femmina e maschio .
La prevalenza aumenta con l’età; il tasso è inferiore all ‘ 1% per i pazienti di età inferiore ai 30 anni e del 7% per i pazienti di età pari o superiore a 70 anni .
Attribuito alla produzione ormonale, sebbene la maggior parte dei tumori non funzioni, una produzione leggermente aumentata di alcuni ormoni può essere verificata in almeno il 15% dei casi.
Valutazione ormonale
Adenomi o carcinomi surrenali che secernono cortisolo, feocromocitomi, aldosteronomi e lesioni secretorie degli androgeni sono i tipi di masse surrenali secretorie o funzionanti diagnosticate in modo più ricorrente .
Tumori che producono cortisolo
Questi tumori di solito producono quantità ridotte di cortisolo, che, nella maggior parte dei casi, non sono sufficienti per aumentare l’escrezione di cortisolo libero nelle urine. Sono, tuttavia, in grado di causare la soppressione dell’asse ipotalamo-ipofisi. Di solito, non ci sono manifestazioni correlate alla sindrome di Cushing nei pazienti. Per questo motivo, questa condizione è nota come sindrome di Cushing subclinica o ipercortisolismo subclinico .
È importante evidenziare la differenza tra la sindrome di Cushing subclinica, caratterizzata da un’anomalia biochimica clinicamente non manifestata e la sindrome di Cushing pre-clinica, che è la fase iniziale dello sviluppo della sindrome stessa. L’ipersecrezione subclinica autonoma dei glucocorticoidi (SAGH) è il termine più attuale, proposto per definire una secrezione autonoma di cortisolo da parte di un adenoma surrenale in pazienti senza sintomi della sindrome di Cushing .
Negli adulti, i segni e i sintomi più suggestivi della presenza di ipercortisolismo includono debolezza muscolare prossimale, pletora facciale, perdita degli arti con aumento del grasso nell’addome e nel viso, ampie strisce viola, ematomi senza traumi evidenti e compresse sopraclavicolari .
Tuttavia, a causa di molti sintomi di hypercortisolism, tra di loro, l’ipertensione e il diabete non sono caratteristici, e il grado del loro aspetto clinico è coerente con la variazione percentuale di sovrapproduzione ormonale, l’indicazione precisa della prevalenza di SAGH, saranno sottoposti ai risultati ottenuti con i metodi dei test utilizzati e i criteri per la selezione dei pazienti sintomatici per la conferma della malattia .
Quando si sospetta la sindrome di Cushing, un test di soppressione del desametasone da 1 mg deve essere eseguito durante la notte. Il giorno prima della raccolta del cortisolo sierico che si verifica alle 8 del mattino, il paziente assume 1 mg di desametasone per via orale alle 11 di sera .
È considerato una risposta anormale del cortisolo quando è presentato nell’intervallo da 1,8 a 5,0 mcg/dl, o più specificamente:
A) Se
B) Se tra 1,8 e 5 mcg/dL: risultati indeterminati;
C) > 5 mcg / dL: diagnosi altamente probabile della sindrome di Cushing;
Una soppressione anormale con desametasone è equivalente ad uno screening sospetto, quindi dovrebbe essere confermata dal dosaggio di cortisolo urinario libero di 24 ore, dopo la somministrazione di 8 mg di desametasone durante la notte e il dosaggio sierico di ACTH, in modo che possa essere determinata l’origine della sindrome di Cushing, che di solito si presenta con livelli non
Nel caso della diagnosi della sindrome di Cushing subclinica, vengono utilizzati i seguenti criteri:
-Cortisolo plasmatico > 5 mcg / dL nel test del desametasone da 1 mg senza alcun altro stigma o almeno 2 dei seguenti risultati .
1.ACTH plasma
2.Cortisolo urinario libero in un campione di 24 ore aumentato;
3.Cortisolo sierico > 3 mcg / dL nel test del desametasone da 1 mg (Figura 1).
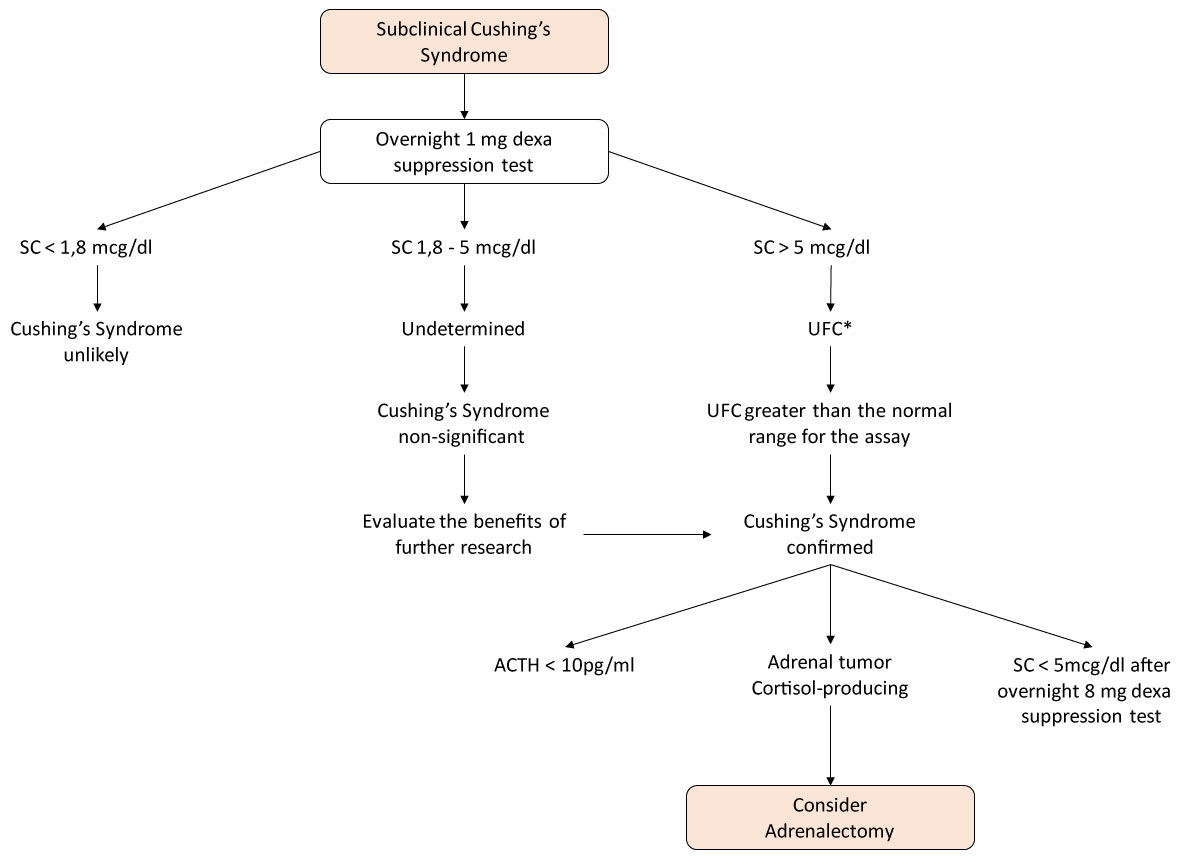 Figura 1: Algoritmo per lo studio della sindrome di Cushing nelle ghiandole surrenali Incidentalomas. * Almeno due misurazioni. Visualizza figura 1
Figura 1: Algoritmo per lo studio della sindrome di Cushing nelle ghiandole surrenali Incidentalomas. * Almeno due misurazioni. Visualizza figura 1
Pertanto, è difficile raggiungere un consenso per l’approccio della sindrome di Cushing subclinica, che può essere trattata clinicamente o attraverso la chirurgia . Per i pazienti con molte comorbidità che possono essere correlate all’ipercortisolismo, il rischio/beneficio dell’surrenectomia deve essere considerato come trattamento. Una gran parte dei pazienti sottoposti a questo intervento chirurgico può sviluppare insufficienza surrenalica acuta (a volte fatale), quindi è estremamente importante la copertura perioperatoria con somministrazione di glucocorticoidi.
Tumori che producono catecolamine
I feocromocitomi sono tumori delle cellule cromaffine del midollo surrenale che producono, immagazzinano, metabolizzano e secernono catecolamine, in alcuni casi, altri ormoni peptidici paragangliomi (PGL) sono tumori simili, ma di origine extra. La sindrome di Feo / PGL è una malattia rara, con una prevalenza stimata tra lo 0,1 e lo 0,2% della popolazione di individui ipertesi .
La maggior parte dei tumori secernenti catecolamine sono sporadici. Tuttavia, alcuni pazienti (circa il 40%) hanno la malattia come parte di un disturbo familiare; in questi pazienti, i tumori secernenti catecolamina hanno maggiori probabilità di essere feocromocitomi surrenali bilaterali o paragangliomi.
Ci sono diversi disturbi familiari associati al feocromocitoma, tutti con ereditarietà autosomica dominante:
• Sindrome di Von Hippel-Lindau (VHL), associata a mutazioni nel gene soppressore del tumore VHL.
• Neoplasia endocrina multipla di tipo 2 (MEN2), associata a mutazioni nel proto-oncogene RET.
• Il feocromocitoma è anche osservato, sebbene raramente, della neurofibromatosi di tipo 1 (NF1), a causa di mutazioni nel gene NF1.
Questi tumori sono di particolare importanza perché, sebbene rari (così come gli adenomi secretori dell’aldosterone) danno origine a una forma di ipertensione correggibile chirurgicamente . L’ipertensione è spesso parossistica. La classica triade di sintomi nei pazienti con feocromocitoma consiste in mal di testa episodici, sudorazione e tachicardia, e vi è una prevalenza di pazienti che non presentano i tre sintomi classici. Oltre ai pazienti con le manifestazioni cliniche tipiche della malattia, l’indagine dovrebbe essere fatta anche in individui con una storia familiare di FEO o carcinoma midollare della tiroide, in presenza di HA in giovane, HA di difficile controllo o induzione anestetica.
La diagnosi di feocromocitoma viene tipicamente effettuata mediante misurazioni di metanefrine e catecolamine fratturate nelle urine e nel plasma, come mostrato nella Figura 2 .
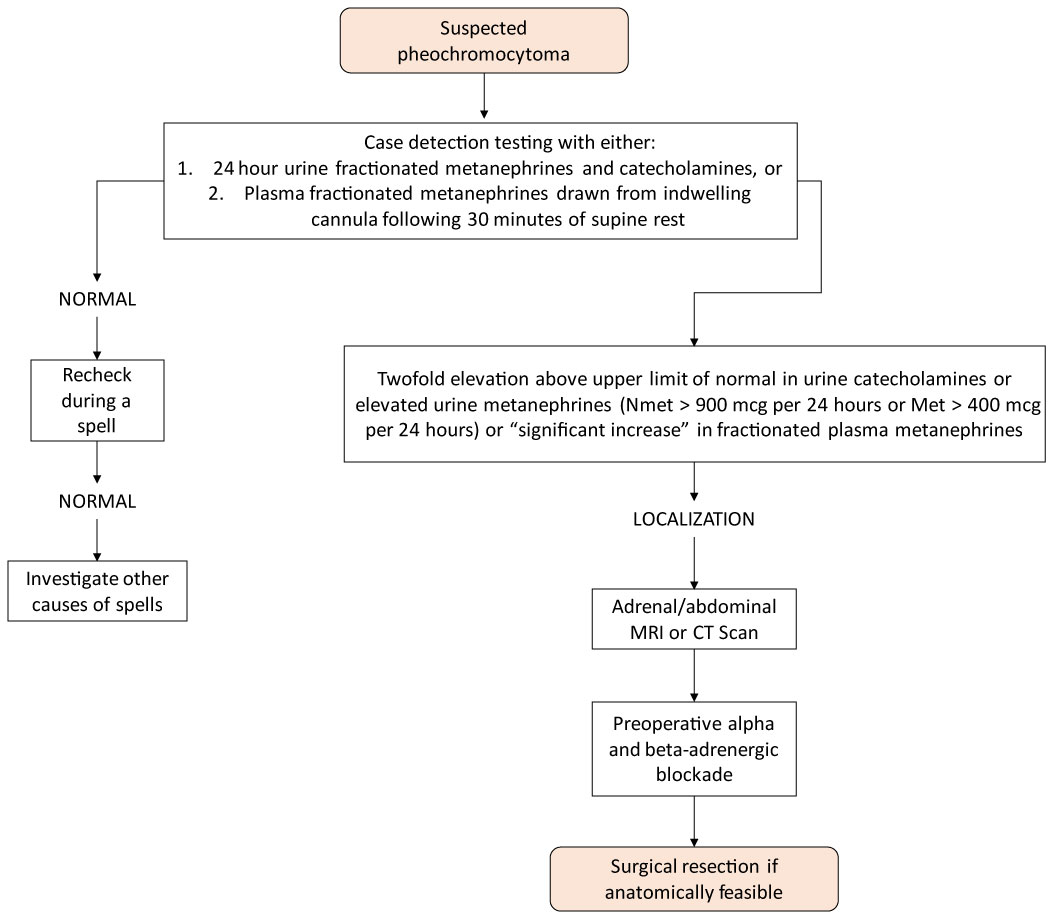 Figura 2: Algoritmo per lo studio dei feocromocitomi nelle ghiandole surrenali Incidentalomas. Vista la Figura 2
Figura 2: Algoritmo per lo studio dei feocromocitomi nelle ghiandole surrenali Incidentalomas. Vista la Figura 2
per Quanto riguarda l’immagine di metodi, Pheochromocytomas ha una maggiore attenuazione su vida CT (> 20 HU), aumento della vascolarizzazione della massa, ritardo di mezzo di contrasto lavaggio (10 minuti dopo la somministrazione di contrasto, un contrasto assoluto medio di lavaggio inferiore al 50%), ad alta intensità del segnale di T2-weighted MRI, cistica emorragica e modifiche e dimensione variabile e può essere bilaterale.
Al contrario, le caratteristiche di imaging che suggeriscono carcinoma surrenalico o metastasi includono: Forma irregolare, densità disomogenea, alti valori di attenuazione CT non potenziati (> 20 HU), dilavamento del mezzo di contrasto ritardato (ad esempio, 4 cm e calcificazione tumorale .
Tumori che producono aldosterone
Gli aldosteronomi sono rari e difficili da rilevare e sono clinicamente caratterizzati da ipertensione arteriosa sistemica correlata all’ipopotassiemia. Considerando che la maggior parte dei pazienti sono asintomatici per quanto riguarda l’ipocalcemia, cioè sono normocalemici, si raccomanda di valutare tutti i pazienti con ipertensione associata a incidosi surrenalica misurando la loro concentrazione plasmatica di Aldosterone e renina Attività plasmatica e il rapporto tra loro superiore a 30 è altamente indicativo di produzione autonoma di aldosterone. Una ragione maggiore di 50 distingue chiaramente l’aldosteronismo primario da altre forme di ipertensione essenziale già un risultato inferiore a 20 contesta la diagnosi e si traduce nell’intervallo tra 20 e 30, indica una diagnosi più accurata.
I pazienti che usano spironolattone non possono essere valutati dalla relazione aldosterone e dall’attività plasmatica. Altri farmaci che possono contribuire a risultati dubbi sono beta-bloccanti, agonisti alfa-adrenergici centrali e antinfiammatori. Allo stesso modo, alcuni farmaci possono mostrare una riduzione dell’inibitore dell’enzima dell’angiotensina conservativa, come il bloccante del recettore dell’aldosterone, gli inibitori della tiazina e la diidropiridina del canale del calcio Figura 3 .
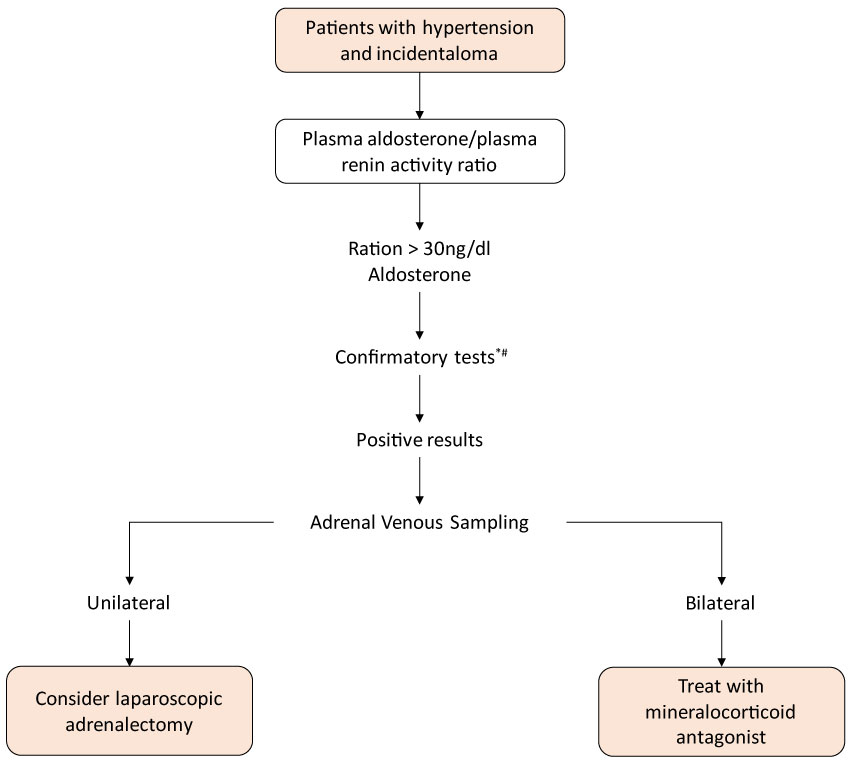 Figura 3: Algoritmo per lo studio dell’iperaldosteronismo negli incidentalomi delle ghiandole surrenali. * Test di conferma più comunemente utilizzati: test di carico di sodio orale, test di infusione salina endovenosa. Visualizza figura 3
Figura 3: Algoritmo per lo studio dell’iperaldosteronismo negli incidentalomi delle ghiandole surrenali. * Test di conferma più comunemente utilizzati: test di carico di sodio orale, test di infusione salina endovenosa. Visualizza figura 3
Il cateterismo surrenale è il metodo di valutazione favorevole alla verifica se l’aumento della produzione di aldosterone in pazienti di età superiore a 40 anni, con iperaldosteronismo confermato, è effettivamente causato da incidentaloma o iperplasia surrenalica.
In tali casi, l’adrenalectomia non risolverebbe l’iperproduzione ormonale, che dovrebbe essere tenuta sotto controllo con l’uso di farmaci, antagonisti dell’aldosterone come lo spironolattone .
Nei pazienti con ipopotassiemia spontanea, renina plasmatica al di sotto dei livelli di rilevamento più aldosterone plasmatico > 20 ng/dl, si suggerisce che non vi è alcuna necessità di ulteriori test di conferma.
Androgeni ed estrogeni producendo tumori
Nei casi di iperplasia surrenalica congenita a causa di un deficit di 21-idrossilasi, è comune trovare masse surrenali, uni o bilaterali, come conseguenza dell’eccessiva stimolazione cronica delle ghiandole surrenali da parte dell’ACTH .
L’ormone sessuale che produce adenomi surrenali è molto raro. Anche i carcinomi che producono androgeni sono rari. I casi di androgeni o estrogeni in eccesso sono raramente descritti in pazienti con adenomi adrenocorticali benigni, ma, in generale, si manifestano con sintomi o segni di virilizzazione nelle donne (acne, irsutismo) o femminilizzazione negli uomini (ginecomastia). Pertanto, tali lesioni non possono essere considerate come veri IAs. Pertanto, la necessità di misurare gli ormoni sessuali e i precursori steroidei è limitata nei casi di lesioni surrenali con indeterminatezza o sospetto di caratteristiche di imaging di malignità, dove livelli elevati possono indicare l’origine adrenocorticale del tumore e suggerire la presenza di un adenocarcinoma surrenale . L’adrenalectomia è indicata per il controllo degli ormoni in individui con virilizzazione o alte concentrazioni di androgeni .
I tumori che producono estrogeni sono rari e, nella maggior parte dei casi, maligni. La presenza di questi tumori negli uomini si manifesta solitamente attraverso la femminilizzazione con ginecomastia, diminuzione della libido, atrofia dei testicoli; nelle donne, può manifestarsi attraverso la tenerezza del seno e il sanguinamento . In questi casi, può anche essere indicata l’adrenalectomia.
Discussione
Gli studi investigativi sull’imaging delle ghiandole surrenali sono di particolare rilevanza per la comprensione che il professionista di quest’area è attento e curioso di qualsiasi tratto di anomalia che sospetti la salute delle ghiandole surrenali.
In parallelo, possiamo osservare l’importanza della conoscenza dei metodi efficaci per la diagnosi e per il trattamento pertinente a ciascun caso rilevato.
Di tutti i protocolli utilizzati e proposti per l’indagine ormonale, quello con la più alta recidiva è quello che utilizza cortisolo sierico dopo sospensione notturna, con 1 mg di desametasone (per ipercortisolismo subclinico) e metanefrine plasmatiche (per il FEO). Lo studio degli aldosteronomi è indicato solo per i casi di ipertensione e o ipopotassiemia, la determinazione dell’aldosterone plasmatico e dell’attività della renina plasmatica.
Numerosi studi pubblicati e diffusi in simposi medici menzionano diversi protocolli utilizzati per l’indagine ormonale, l’osservazione e la scoperta di anomalie delle cellule tissutali delle ghiandole surrenali, i diversi tumori che queste ghiandole possono ospitare e misure preventive e terapeutiche delle malattie, offrendo qualità della vita ai pazienti.
Riconoscimenti/Conflitti di interesse
Nessuno degli autori ha un conflitto di interessi.
Citazione
do Prado BC, Schafascheck GS, Puppim AR (2019) Valutazione ormonale dei tumori surrenali: ciò che il medico generico comune dovrebbe sapere. Int Arch Urol Complic 5: 063. doi.org/10.23937/2469-5742/1510063