MAE. Det finns många distinkta barndomsepilepsisyndrom som vi har blivit kritiskt medvetna om i den genomiska eran eftersom de är kopplade till framstående genetiska orsaker, inklusive Dravet syndrom (SCN1A) och epilepsi av spädbarn med migrerande fokala anfall (KCNT1). Det finns dock många andra epilepsisyndrom där en genetisk orsak länge har misstänkts, men har förblivit svårfångad. Ett av de epilepsisyndrom som till stor del har förblivit outforskat är Doose syndrom, även kallad myoklonisk Astatisk epilepsi (MAE) eller epilepsi med myoklonisk-atoniska anfall. I en ny studie i epilepsi undersökte vi den genetiska arkitekturen av Doose syndrom och identifierade monogena orsaker hos 14% av individerna, inklusive SYNGAP1, NEXMIF (KIAA2022) och SLC6A1. Vår studie tyder på att Doose syndrom är genetiskt heterogent, eventuellt med ett distinkt genetiskt landskap.
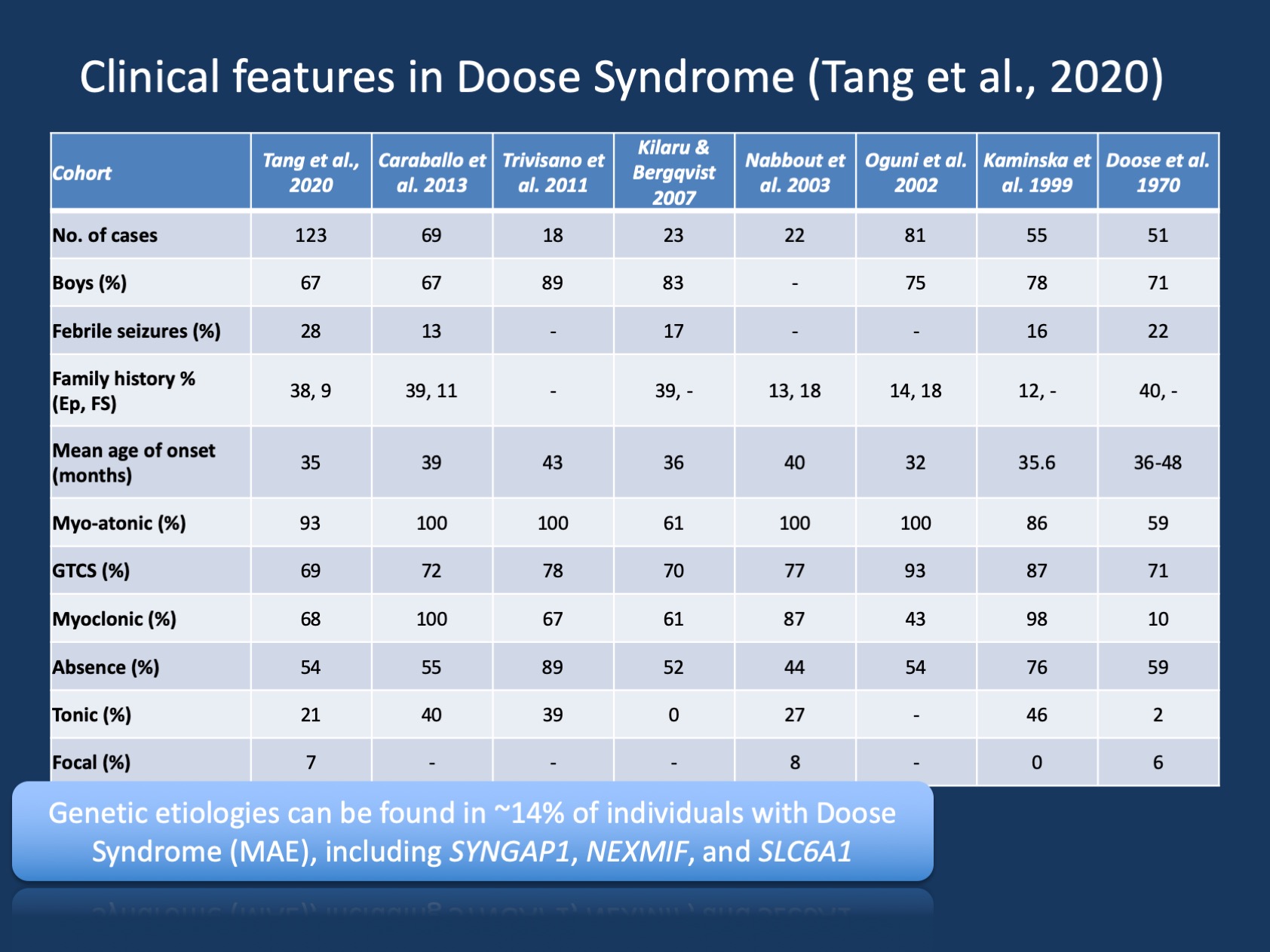
Figur 1. De kliniska egenskaperna hos Doose syndrom (epilepsi med myokloniska atoniska anfall eller myoklonisk Astatisk epilepsi ). Tabellen är anpassad från vår senaste publikation av Tang et al., 2020 och jämför de kliniska egenskaperna i de stora kohorterna av Doose syndrom som har rapporterats sedan den ursprungliga beskrivningen 1970. Doose syndrom (MAE) är ett sällsynt epilepsisyndrom, men är en igenkännlig klinisk enhet. I motsats till Dravet syndrom där >90% av individerna har sjukdomsframkallande varianter i SCN1A är Doose syndrom genetiskt heterogent.
historien om MAE. Först och främst, låt mig rensa upp några namnproblem. Doose syndrom eller myoklonisk Astatisk epilepsi (MAE) kallas nu officiellt epilepsi med myoklonisk-atoniska anfall med den senaste namngivningskonventionen. Men medan vi vanligtvis försöker anpassa oss till det officiella sättet för hur epilepsirelaterade termer används, Låt mig vara lite envis här. Som barnneurolog utbildad vid Institutionen för Neuropediatri i Kiel, Tyskland, kommer jag att hänvisa till detta tillstånd som Doose syndrom med MAE som en stenografi och hedra Hermann Doose (1927-2018) som är en av grundarna av europeisk pediatrisk epileptologi (länk till ILAE in memoriam). Många engelska som modersmål får vanligtvis uttalet fel-det korrekta uttalet är ”Dohs-ah” och rimmar inte med ”gås”.
den kliniska bilden av MAE. Doose krediteras vanligtvis med att göra en viktig klinisk observation: på 1960-talet erkände han och hans medarbetare i Kiel en ovanlig epilepsi hos barn som kännetecknades av plötslig början av droppattacker (atoniska anfall), myokloniska anfall, generaliserade tonisk-kloniska anfall och ett generaliserat EEG-mönster, som vanligtvis förekommer mellan två och fem års ålder. Denna epilepsi inträffade vanligtvis hos barn med tidigare typisk utveckling (se nedan) och resulterade ofta i utvecklingsfördröjning och intellektuell funktionsnedsättning. Doose och medarbetare erkände att detta nyligen erkända epilepsisyndrom skilde sig från Lennox-Gastaut-syndrom eller epilepsier med generaliserade tonisk-kloniska anfall utfällda av feber, vars senare senare konceptualiserades som Dravet-syndrom.
förstå MAE. Kliniska beskrivningar av Doose syndrom (MAE) har varit stabila under åren, men MAE har inte fått så mycket uppmärksamhet som barndomsepilepsi syndrom där en genetisk grund hade upptäckts och där riktad forskning kan möjliggöra nya behandlingar. I vår senaste publikation av Tang och medarbetare har vi listat åtta studier under de senaste 50 åren mellan 1970 och 2020 med totalt 442 beskrivna individer. Följaktligen är MAE sällsynt, men det är vanligt att de flesta barnneurologer och pediatriska epileptologer är medvetna om detta tillstånd. Endast en liten delmängd av individer med MAE har strukturella hjärnfynd eller metaboliska abnormiteter som förklarar den underliggande epilepsin – hos de allra flesta barn med MAE är neuroimaging och metabolisk upparbetning unremarkable.
hitta orsaken till MAE. Frånvaron av förklarande avbildning eller metaboliska fynd väcker oro för en underliggande genetisk orsak. Medan tidigare studier hade försökt fästa på ett genetiskt bidrag genom epidemiologiska studier genom att bedöma familjehistorier, har denna traditionella metod till stor del ersatts av nya upptäckter inom epilepsigenetik. Att bedöma den genetiska grunden för allvarliga barndomsepilepsier görs nu genom molekylär genetik snarare än genetisk epidemiologi. Att bedöma Mae: s genetiska arkitektur var ett av målen i vår studie av Tang och medarbetare som publicerades i epilepsi.
är MAE en sjukdom? Innan jag beskriver de genetiska fynden i MAE, låt mig presentera en pågående debatt inom MAE-fältet. Den initiala beskrivningen av Doose och medarbetare inkluderade till stor del barn med typisk utveckling före anfall (>75%), vilket är jämförbart med vår senaste studie (79%). Men när man jämför barn med och utan tidigare utvecklingsproblem, tittar vi på samma sjukdom eller bör endast barn med tidigare obekväm utveckling betraktas som typiska MAE? Även om denna fråga inte lätt löses när man överväger epilepsihistorierna och resultaten, kan de underliggande genetiska förändringarna ge några ledtrådar. Genetiska orsaker identifieras oftare hos barn med allvarligare neuroutvecklingsstörningar och alla barn med identifierade genetiska orsaker hade komplexa neuroutvecklingsstörningar utöver epilepsin. Följaktligen kan genetiska orsaker observeras hos barn med MAE, men vanligtvis endast hos barn med MAE och comorbida neuroutvecklingsstörningar, vilket tyder på att MAE inte bara är ett genetiskt heterogent tillstånd utan har liknande egenskaper som annan epilepsi i barndomen med avseende på deras genetiska heterogenitet och komorbiditet. Till exempel i West syndrom (Infantil spasmer), genetiska orsaker identifieras också endast hos barn med neurodevelopmental fynd och inte hos barn som direkt svarar på medicinering utan neurodevelopmental följder.
genetiska fynd i MAE. Det genetiska spektrumet i MAE är annorlunda än i epileptiska encefalopatier i stort. Genetisk testning utfördes hos 85 individer och 12/85 (14%) individer hade positiva genetiska fynd. Tre gener hittades hos två individer, inklusive SYNGAP1, NEXMIF (KIAA2022) och SLC6A1. Denna triad av gener har tidigare kopplats till utvecklings-och epileptiska encefalopatier med generaliserade anfall. Dessutom hittades enskilda individer med sjukdomsframkallande varianter i KCNA2, SCN2A, STX1B, KCNB1 och MECP2. Alla dessa gener diskuterades tidigare på vår blogg och är kända för att presentera generaliserade anfall och generaliserade EEG-funktioner.
frånvarande gener i MAE. Medan vissa tidigare rapporter har hittat varianter i SCN1A, hittades detta inte i vår kohort. Dessutom är andra vanliga epilepsigener särskilt frånvarande, inklusive CDKL5 och SLC2A1 (GLUT1). Detta tyder på att Mae: s genetiska landskap är något distinkt och skiljer sig från andra utvecklings-och epileptiska encefalopatier, även om endast begränsade slutsatser kan dras från relativt små kohorter. Det diagnostiska utbytet i MAE är inte särskilt högt-14% är mycket lägre än vad som vanligtvis ses i utvecklings-och epileptiska encefalopatier. Men när man utesluter individer med typisk utveckling är de diagnostiska utbytena mycket högre, vilket tyder på att genetisk testning är av värde hos barn med MAE. I allmänhet, även om ingen enskild individ med MAE hade sjukdomsframkallande varianter i SLC2A1, kan genetisk testning för behandlingsbara orsaker till genetiska epilepsier i allmänhet övervägas i atypiska generaliserade epilepsier.
den saknade ärftlighet MAE. Om genetisk testning är negativ för MAE hos mer än 80% av individerna, var är den dolda genetiska bördan? Från och med 2020 föreslår jag två möjliga förklaringar. För det första är den genetiska bördan av generaliserade epilepsier inte helt okänd. Mer än 30% av populationsrisken för tillstånd som epilepsi i frånvaro av barn (CAE) eller juvenil myoklonisk epilepsi (JME) förklaras. Denna förklaring ges emellertid inte av orsaker med en gen, utan av polygenisk risk, med hänvisning till additiv effekten av tusentals vanliga genetiska varianter. Det kan antas att MAE kan representera en” extrem fenotyp ” av generaliserade epilepsier där denna polygena risk är särskilt hög. Denna hypotes är testbar och kommer definitivt att fortsätta i framtiden. Dessutom finns det mer i det mänskliga genomet än exomen – nya fynd i epilepsierna har betonat rollen som icke-kodande varianter, såsom upprepade utvidgningar i familjär vuxen myoklonisk epilepsi (berömmelse). Med tanke på de slående likartade kliniska egenskaperna hos många barn med MAE kan det fortfarande vara rimligt att leta efter en vanlig genetisk orsak för många barn med MAE utanför exomen, med hjälp av hela genomsekvensering. Att tillsammans bedöma polygena riskpoäng (PRS) och hela genomsekvensering (WGS) är två potentiella vägar för att ytterligare identifiera den genetiska grunden för MAE.
vad du behöver veta. Doose syndrom (MAE) är ett förmodligen genetiskt epilepsisyndrom med en heterogen orsak. Sjukdomsframkallande varianter förekommer hos 14% av barnen och identifieras vanligtvis hos barn med ytterligare neuroutvecklingsfunktioner som utvecklingsfördröjning eller autism. En triad av gener inklusive SYNGAP1, NEXMIF (KIAA2022) och SLC6A1 är återkommande genetiska orsaker som kan antyda en gemensam underliggande biologi som skiljer sig något från andra utvecklings-och epileptiska encefalopatier.

Ingo Helbig är barnneurolog och epilepsigenetikforskare som arbetar på Children ’ s Hospital of Philadelphia (CHOP), USA. Han leder också epilepsigenetikgruppen vid universitetet i Kiel, Tyskland.

